
[ARTICLE] In-Vitro Diagnostic Medical Devices Q&A: State of the Art in IVDR
As required by the IVD Medical Device Regulation (EU) 2017/747, a device performance evaluation plan must contain a description of the state of the art pertaining to the IVD device under evaluation.
All relevant aspects related to the evaluation of the performance of a device must be weighed against the current state of the art. These include:
- the demonstration of scientific validity, device analytical performance and clinical performance;
- the evaluation of the risk control measures implemented;
- the technical safety of the device;
- the IVD’s expected clinical benefit.
In other words, the clinical evidence must be such as to scientifically demonstrate, by reference to the current state of the art of medicine, that the IVD medical device has a positive risk/benefit ratio. Cited 20 times in the IVDR (EU) 2017/747I, for example in relation to the design of in vitro diagnostic medical devices, as well as risk management, performance evaluation and post-market surveillance, it is clear that state-of-the-art assessment plays a central role in the overall life cycle of a device and not just in relation to performance evaluation.
In this blog post, we provide comprehensive answers to some of the most common questions about the state of the art in IVD medical devices.
What does “State of the Art” mean?
Manufacturers are expected to regularly, throughout the device life cycle, review and evaluate the evolution of the state of the art for their IVD medical devices.
However, there is no formal definition of state of the art in the IVD regulations. MDCG 2022-2 published in February 2022 and entitled “Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs)” defines state of the art as:

It further clarifies that the state of the art embodies what is currently and generally accepted as good practice in technology and medicine, which does not necessarily represent the most technologically advanced solution.
Therefore, state of the art should be understood as the accepted standard clinical practice in a particular medical field, which is not necessarily the most advanced technology, and should be used to demonstrate the acceptability of the IVD medical device based on that current knowledge.
As per IVDR requirements, the state-of-the-art description shall include the identification of existing relevant standards, CS, guidance or best practices documents. Furthermore, the state-of-the-art assessment may contain a mix of:
- what is currently considered best clinical practice for diagnosing/detecting or monitoring a particular condition/disease;
- what biomarkers are commonly accepted;
- what technology is most commonly used to measure those biomarkers.
As the state of the art evolves over time, such an evaluation must be conducted regularly to reflect the most recent knowledge.
To give a concrete example of the possible evolution of the state of the art over time: for the diagnosis of myocardial infarction, the creatine-kinase-MB assays were for years considered the standard of care. In the last 10 years, however, these assays have been replaced in routine clinical practice by assays measuring cardiac troponin and high-sensitivity cardiac troponin. However, the technology for detecting these biomarkers, namely immunoassays, remains the same.
When to start the assessment of the state of the art?
The findings of the state-of-the-art assessment will impact several parts of the device's technical documentation. First, the results of the state-of-the-art assessment may trigger design choices, such as using one particular technology over another or using a specific biomarker or set of biomarkers over others.
In addition, the risk control measures adopted by manufacturers for the design and manufacture of devices must be in line with safety principles, taking into account the generally accepted state of the art. The results of the state-of-the-art assessment will therefore also have an impact on risk assessment and management.
Furthermore, the "state of the art" is not only used to address the clinical evidence of the device, thus being part of the performance evaluation plan, but it is also used to address the safety and performance summary for Class C or Class D devices and is a key component of the post-market surveillance (PMS) requirements. State-of-the-art surveillance is considered a proactive PMS activity and must be covered in the PMS plan.
Therefore, a state-of-the-art assessment must be conducted very early during the development of the device and must be regularly reassessed and updated, when necessary, through the PMS activities and during the entire life cycle of the device. The design and risk/benefit ratio of the device should be considered in light of the results of the most recent state-of-the-art evaluation.
How should the state of the art be assessed?
The assessment of the state of the art involves a systematic and methodological approach to the literature review, which requires time and resources.
Acceptable inputs for assessing the state of the art for an IVD may include:
- Published common specifications such as those published for the assessment of the performance of SARA-CoV 2 assays and for other class D devices
- Standards used for the same or similar devices
- Clinical guidance documents and medical textbooks
- Best practices, as used in other devices of the same or similar type
- Expert consensus documents produced by professional medical associations
- Publications from regulatory authorities or additional information for similar other products
- Recommendations from medical or laboratory associations
- Comparison of the benefits and risks of the device under development with the benefits and risks of similar devices available on the market
- Peer-reviewed literature, such as review articles on relevant condition/disease managed by the device, biomarkers or device technology.
The assessment of the literature must be conducted using explicit and systematic data appraisal and collection. For IVD medical devices there are currently no specific guidance documents describing how such a methodological review of the literature needs to be conducted to achieve an adequate state-of-the-art assessment.
The guidance document MEDEV 2.7/1 rev 4 entitled "Clinical Evaluation: A Guide for Manufacturers and Notified Bodies under Directives 93/42/EEC and 90/385/EEC" provides standards of good practice for methodological literature reviews, offering general instructions on literature searching and describing methods for developing a protocol for conducting a literature review.
Although this guidance document does not address in vitro diagnostic devices per se, it nevertheless represents the gold standard for methodologically reviewing the state of the art and can be used by manufacturers of in vitro diagnostic devices for this purpose.
It is important that manufacturers document the databases used for the literature search, as well as the applied search strings and keywords, and that identified articles are appraised using traceable and systematic inclusion/exclusion criteria.
Conclusion
In conclusion, the assessment of the state of the art is essential because it is needed to confirm the clinical benefit of the device, to justify the positive risk/benefit ratio of the IVD, and also to guide design choices and risk mitigation.
State-of-the-art evaluation is a living concept, which means that it must be evaluated throughout the life cycle of the device, with the clinical benefit of the device always being updated in light of the most recent state-of-the-art evaluation.
Most manufacturers are aware of the latest state of the art regarding their IVDs, but this is not always documented in a systematic and methodological way in the device's technical documentation. This can lead to unnecessary delays and an additional burden on resources when the device undergoes conformity assessment by a Notified Body.
It is therefore recommended that a methodological assessment of the state of the art be performed and documented early in the life cycle of the device and updated regularly to reflect the latest knowledge in the relevant medical field.
How Medidee can help
Medidee’s IVDR experts collaborate with your team to design and implement a strategy that is tailored to your specific challenges and needs, for a well-managed transition to IVDR.
Contact us to get started now!
This article was written by Dr Silvia Anghel and Dr Julianne Bobela.

[ARTICLE] International Market Access Strategy for Electrical Medical Devices and IVDs: National Differences and the Added Value of "CB Scheme" and "NRTL Listing Report"
This blog article focuses on the international market access strategy for medical electrical equipment, notably electrical medical devices including in vitro diagnostic medical devices (IVDs) and explains the relevance and benefits of establishing an IEC 60601 or IEC 61010 test report and certificate, following the CB (Certification Body) scheme test procedure, and respecting the national differences. In addition, the purpose of NRTL (Nationally Recognized Testing Laboratory) listing report along with its requirements and benefits are summarized.
Manufacturers of electrically-driven medical devices and IVDs may face different regulatory requirements depending on the intended device market strategy. Due consideration of the applicable international market requirements during and after the development phase of the medical electrical equipment is therefore essential.
As part of the global market access strategy for medical electrical equipment, medical device and IVD manufacturers must comply with the “State-of-the-Art” (SOTA) concept. This is normally achieved through the application of relevant harmonized standards within the European Union, or of Food and Drug Administration (FDA) recognized standards for the United States of America, taking into consideration the device classification and intended use of the device/system.
General Considerations on International Market Access for Medical Electrical Devices and IVDs
First of all, medical device manufacturers should begin by determining the target countries in which they intend to place their medical devices on the market. Based on the country, it should be assessed if any national differences are applicable. Identified national differences should be considered early during the development of the medical device. This approach, known as “frontloading”, can enable substantial cost savings compared to conducting this process at an advanced development stage.
For instance, IEC 60601-1:2005+AMD1:2012+AMD2:2020 (3.2 Edition) and IEC 61010-1:2010+AMD1:2016 (3.1 Edition) contain a variety of national differences in applicable regulatory requirements.
Notably, the following country-specific IEC 60601-1 versions are applicable depending on the target country and type of medical electrical equipment at stake:
- ANSI/AAMI ES60601-1:2005/A2:2021 (United States)
- CAN/CSA-C22.2 NO.60601-1:14 (Canada)
- SI 60601 Part 1 (Israel)
- JIS T 0601-1:2017 (Japan)
- MFDS No. 2020-12 (Republic of Korea)
- BS EN 60601:2006 A1 (United Kingdom)
The following country-specific differences of IEC 61010-1 are applicable depending on the target country and type of in vitro diagnostic:
- CAN/CSA-C22.2 NO.61010-1+Amd1 (Canada)
- EN 61010-1:2010/A1:2019 (CENELEC)
- UL 61010-1 (3rd) AM.1 (United States)
The identified applicable national differences based on the target markets should be considered during the development and testing (verification) of electrical medical devices and IVDs.
It is important to note that FDA only recognizes the ANSI AAMI ES60601-1 standard, meaning that if the medical electrical equipment manufacturer plans to obtain market approval/clearance in the USA, they need to comply with the national differences for the USA presented therein.

Figure 1: Screenshot of ANSI AAMI ES60601-1 of FDA-recognized standard database
Furthermore, manufacturers should bear in mind that ANSI AAMI ES60601-1 3.1 Edition will be superseded by 3.2 Edition by 17 December 2023 according to the information provided by FDA, and therefore that after that date FDA will only accept submissions complying with 3.2 Edition.
With regard to the 61010-1 standard, the FDA recognizes the ANSI UL 61010-1 3rd Edition and IEC 61010-1 Edition 3.1.

Figure 2: Screenshot of ANSI UL/ IEC 61010-1 of FDA-recognized standard database
In a second step, medical device and IVD manufacturers should contact an external test laboratory to plan and conduct the required tests (e.g., as per IEC 60601-1 or IEC 61010-1) in accordance with the identified applicable national differences.
As an example – Canadian national difference (CAN/CSA-C22.2 No. 60601-1:14) requires, as stated in clause 8.6.4 “impedance and current-carrying capability”, compliance to CSA C22.2 No. 04. This implies a protective earth test current for cord-connected equipment of twice the rating of the attachment plug cap, or for permanently installed equipment, of twice the rating of the fuse for branch circuit, but not less than 40 A for 120 seconds.
In contrast, IEC 60601-1 3.2 Edition requires, according to clause 8.6.4 “impedance and current-carrying capability”, a protective earth test current of 25 A or 1.5 times the highest rated current of the relevant circuit for a maximum time of 10 seconds.
The Canadian national difference requirement in the above example is therefore stricter than the IEC 60601-1 3.2 Edition requirement. Depending on whether the equipment is cord-connected or permanently installed, the extent of the required construction (protective earth wire diameter) and compliance testing could be impacted.
Finally, please note, IECEE covers 23 categories of electrical equipment and testing services. Therefore, the CB Scheme is not limited to electrical equipment for medical use. As an example, three additional categories of electrical equipment are listed below, which are often used/ tested in combination with electrical medical devices and IVDs:
- Electromagnetic Compatibility (EMC)
- Safety Transformers and similar equipment (SAFE)
- Information Technology Audio Video (ITAV)
If it is intended to gain market access internationally, it is recommended to follow the IECEE (IEC System of Conformity Assessment Schemes for Electrotechnical Equipment and Components) CB (Certification Body) scheme of test procedure which facilitates the global market access.
What is the “CB scheme”?
The CB scheme is an international system for mutual acceptance of test reports and test certificates. Currently, 54 countries are member bodies taking part in CB scheme approach. Only certification body testing laboratories (CBTL) may issue CB test reports and certificates. All CBTLs can be viewed here.
Typically, the first page of an IEC 60601/ IEC 61010 test report indicates the test specification and the applied test procedure.

Figure 3: Example of IEC 60601 test report test specification
What are the implications for electrical (IVD) medical device manufacturers?
The CB scheme follows predefined requirements, and the medical device manufacturer and the external test laboratories are obliged to comply with specific rules, operational documents, and guidance.
For instance, the IECEE Operational Document OD-2055 Annex D requires IEC 60601-1:2005+A1:2012+A2:2020 compliance, in addition to a test report documenting compliance of usability in accordance with IEC 60601-1-6:2010+A1:2013+A2:2020. Any other applicable collateral standard (e.g., IEC 60601-1-3) shall also be considered.
Moreover, it must be noted that following the CB scheme does incur additional costs due to the additional requirements and certification fees (certificate) implied.
Last, it is important to know that according to clause 6.2.1 of IECEE OD-2020, only three amendment reports due to technical changes are accepted to the original issued test report and test certificate. After three amendments a new CB test report and certificate need to be requested and issued by the external test laboratory. Even upgrading the CB test certificate and report from an old edition to a newer edition of the standards requires a new CB test report and certificate.
Besides that, the CB scheme offers the following benefits to the medical device manufacturer:
- It enables fulfilment of the criteria for obtaining market access in certain countries – it provides direct acceptance by the regulatory authorities in many countries and is accepted by numerous retailers, buyers and vendors worldwide
- No additional duplicate testing is needed, and it is a globally approved and well-established procedure
- Simplification of the roll-over process (changing the external test laboratory)
- All CB certificates are listed in the IECEE database and are publicly accessible
Now, moving on to the NRTL listing report:
What is the “NRTL”?
Nationally Recognized Testing Laboratory (NRTL) are mainly U.S.-based test laboratories recognized by the OSHA (Occupational Safety Health Administration) according to the Code of Federal Regulation (CFR) 21 §1910.7 to perform certification for certain products (e.g., medical electrical equipment or in vitro diagnostic) to ensure they meet the constructional and general industry OSHA standards. The NRTL will issue a separate listing report for the medical electrical equipment/ in vitro diagnostic manufacturer.
OSHA requires NRTL testing and affixture of the NRTL safety mark for electrical products used in public buildings or workplaces (e.g., hospital, clinics, and laboratories) in the United States. This is regulated by the National Electric Code (NEC) §90.7, §110.2, and §110.3 (C), and may be subject to control by Authorities Having Jurisdiction (AHJ) during the installation of the medical electrical equipment/ in vitro diagnostic or during operation.
What are the implications for electrical (IVD) medical device manufacturers?
- Electrical (IVD) medical device manufacturers require an external NRTL-recognized test laboratory. Currently, there are 21 listed NRTL-recognized laboratories
- The NRTL listing report and certificate need to be requested, incurring additional costs
- Initial factory inspection must be conducted by an NRTL inspector, and follow-up inspections at the factory site are needed every 3 or 6 months (additional initial and follow-up costs incurred)
- Manufacturers need to update the NRTL mark on the type label, and within the technical documentation such as in the instructions for use
The NRTL listing report puts focus on general constructional requirements (cable routing, labeling, protective earth connection, list of critical components) and routine tests for manufacturing and production (grounding continuity test, dielectric voltage withstand test, and leakage current test). These aspects will be regularly inspected during the ongoing factory inspections.
In addition, if it is also intended to obtain market access in Canada, it is strongly recommended to directly apply for an NRTL listing report for Canada in parallel. The NRTL listing report approach requested by the Standards Council of Canada (SCC) is similar to that for the U.S., and one combined NRTL listing report can be established for both countries.
Besides that, the NRTL listing report offers the following benefits to electrical (IVD) medical device manufacturers:
- It enables market access for the United States of America and Canada
- The electrical (IVD) medical device will bear the NRTL mark, showing that the NRTL tested and certified the product (quality property)
- Valid NRTL-certified medical electrical equipment, including electrical (IVD) medical devices, are listed in the related NRTL database
How can Medidee, now part of Veranex, support your company?
Our team of electrical safety specialists at Medidee can assist you in identifying the regulatory requirements, notably the applicable standards and national differences, for your target market(s). Moreover, Medidee can help you in determining if a test report according to the CB scheme and/ or NRTL listing report is mandatory in specific countries. In addition, we can support you in selecting the right external test laboratory depending on the intended global market access strategy and during the certification process and on factory inspection to ensure a smooth certification process of the medical electrical equipment or in vitro diagnostic.
Contact us to discuss your needs.
This article was written by Stefan Staltmayr and Dr Jérôme Randall.
[ARTICLE] Combination Products: Similarities and Differences of EU and US Regulations
The success of novel combination products is much dependent on the regulatory intelligence and foresight to define the best strategy to place and maintain them on the market, through a deep understanding of how drugs, devices and their combination are regulated in the different target markets.
This article aims to provide general guidelines for combination product manufacturers who may not be familiar with the EU and US regulations, or know one of these markets well and aim to access the other.
Terminology and definitions
The term “combination product”, commonly used to identify the category of medical products made of a combination of drug, device, and/or biologic, comes from the FDA definition given in 21 CFR 3.2 (e).
In the EU, instead, the wording “integral product” is used to refer to devices incorporating a medicinal substance (Article 1 §8), and devices intended to administer a medicinal product (Article 1 §9), as defined in the Medical Device Regulation (EU) 2017/745 (MDR). Of note, the European Medicines Agency (EMA) has published a Q&A guidance document on this topic and the term “Drug Device Combination” (DDC) is also used.
In 21 CFR 3.2 (e), the FDA defines the following types of combination products:
- single-entity combination product in which its constituent parts are physically, chemically, or otherwise combined or mixed and produced, as a single entity;
- co-packaged combination product in which its constituent parts are packaged together in a single package or as a unit;
- cross-labeled combination product in which its constituent parts are packaged separately and, according to the investigational plan or proposed labeling, are intended for use together to achieve the intended use, indication, or effect.
According to the MDR, the same products are categorized differently, as follows:
- devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device;
- devices incorporating a medicinal substance as an integral part which has an action principal to that of the device;
- devices intended to administer a medicinal product, placed on the market as a single integral product (i.e. placed on the market in such a way that they form a single integral product which is intended exclusively for use in the given combination and which is not reusable);
- devices intended to administer a medicinal product, placed on the market as a “non-integral product”.
To provide some examples, a pre-filled syringe is a single-entity combination product in the US, and “a device intended to administer a medicinal product as a single integral product” in the EU.
A drug-eluting stent is a single-entity combination product in the US, and “a device incorporating a medicinal substance as an integral part which has an action ancillary to that of the device” in the EU.
In the US, insulin cartridges to be used with a reusable pen injector can be a co-packaged combination product, if the insulin cartridges are provided in the same package with the reusable pen, or a cross-labeled combination product, if provided separately. In the EU, the reusable pen injector is “a device intended to administer a medicinal product as a non-integral product”.
Mode of Action to define how the product is regulated
Interestingly, the term “mode of action” is used both in the US and in the EU, but its definition is provided only in 21 CFR 3.2(k), which states that the “mode of action is the means by which a product achieves an intended therapeutic effect or action”.
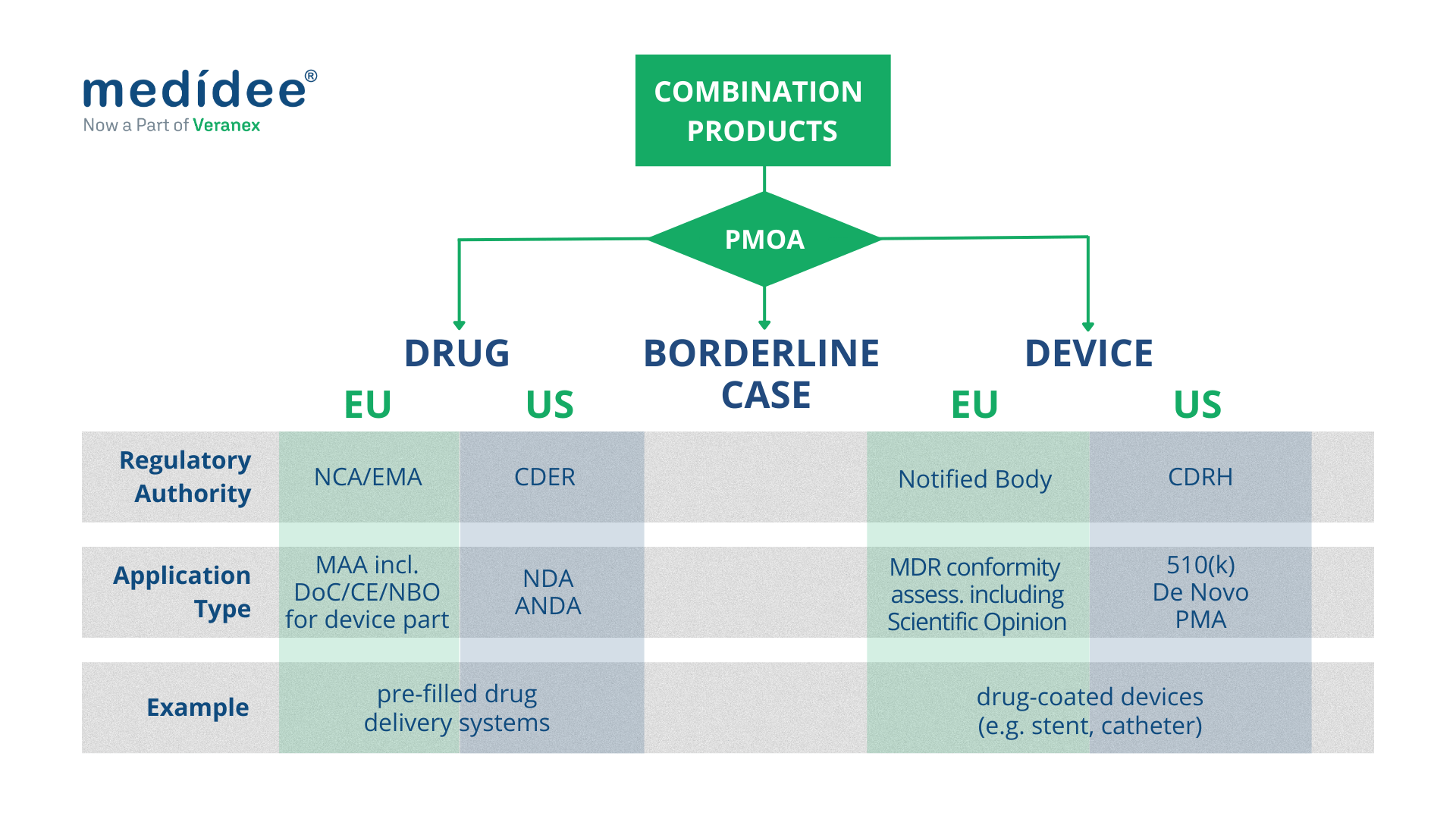
Both in the US and in the EU, combination products are recognized to have more than one identifiable mode of action. Among the multiple modes of action, FDA defines as Primary Mode of Action (PMOA) “the single mode of action of a combination product that provides the most important therapeutic action of the combination product. The most important therapeutic action is the mode of action expected to make the greatest contribution to the overall intended therapeutic effects of the combination product.” (21 CFR 3.2(m)).
The MDR does not provide a definition, but rather distinguishes between “principal” and “ancillary” action of the medicinal substance to that of the device.
Finally, the “primary” or “principal” mode of action is the key criterion used in both US and EU's regulatory frameworks to define how the product is regulated and by which Regulatory Authority.
Combination Products regulated as medical devices
Both in the EU and in the US, combination products with a device PMOA, also called device-led combination products, are regulated as medical devices.
European Union
In the EU, this type of combination product includes “devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device”, and “devices intended to administer a medicinal product, placed on the market as a non-integral product”. They undergo conformity assessment procedures as any other medical device, as defined in Article 52 and set out in Annex IX to XI of the MDR.
In terms of classification, “devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device” are class III medical devices, according to Rule 14, as defined in Annex VIII of the MDR.
Instead, “devices intended to administer a medicinal product, placed on the market as a non-integral product” are classified differently depending on their intended purpose. Of note, the MDR up-classified some of these products: for example, inhalers were regulated under the Medical Device Directive 93/42/EEC (MDD) as class I devices, whereas they are now class IIa or class IIb devices under the MDR.
Finally, as part of the conformity assessment procedure, “devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device” require consultation by the Notified Body of the relevant Medicinal Products Authority (National Competent Authority or EMA), to seek their Scientific Opinion. This consultation process should be taken into account when planning the CE marking of such products or when planning a significant change to an already CE-marked product, as it greatly impacts the timelines for completion of the conformity assessment procedure.
United States
In the US, as a general rule, FDA requires a single application to streamline regulatory interactions with the Agency and the marketing application type generally coincides with the PMOA of the combination product. Hence for a device-led combination product, a single application should be submitted to the Center for Devices and Radiological Health (CDRH) which acts as “lead center”.
A Premarket Approval (PMA) is required for class III device-led combination products; a De Novo Classification Request applies for novel device-led combination products for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use (class I and class II); a Premarket Notification (510(k)) applies when the substantial equivalence of the device-led combination product can be demonstrated against a predicate product.
Of note that, as part of the single application, sufficient information should be provided on the drug constituent part and may include nonclinical pharmacology, toxicology and clinical pharmacology (including pharmacokinetic) data and chemistry, manufacturing, and controls (CMC) information.

Combination Products regulated as drugs
Both in the EU and in the US, combination products with a drug PMOA, also called drug-led combination products, are regulated as drugs.
European Union
In the EU, “Devices incorporating a medicinal substance as an integral part which has an action principal to that of the device” and “devices intended to administer a medicinal product, placed on the market as a single integral product” are regulated as medicinal products under Directive 2001/83/EC or Regulation (EC) No 726/2004, as applicable.
In this context, Article 117 of the MDR introduces an amendment to Directive 2001/83/EC, requiring that the Marketing Authorisation Application (MAA) includes the results of the assessment of the conformity of the device part with the relevant General Safety and Performance Requirements (GSPRs) set out in Annex I of the MDR. This translates into two cases:
- if the device part classifies as a class I medical device, then the evidence of conformity to the relevant GSPRs is provided as part of the manufacturer's EU Declaration of Conformity;
- if the device part classifies in any other way (i.e. class Im, class Is, class Ir, class IIa, class IIb, and class III), then the evidence of conformity to the relevant GSPRs is provided either as the CE certificate issued by a Notified Body (if the device is CE marked), or as the so-called Notified Body Opinion. In the latter case, careful planning of the submission of the Technical Documentation of the device part to the Notified Body must be done to ensure sufficient time for review by the Notified Body and obtain the Notified Body Opinion, to be included as part of the MAA.
United States
In the US, for a drug-led combination product, a single application should be submitted to the Center for Drug Evaluation and Research (CDER) which, acts as “lead center”.
For a new drug-led combination product, the single application is a New Drug Application (NDA); for drug-led combination products that are the generic version of already-approved drug-led combination products, the Abbreviated New Drug Application (ANDA) is, in general, the most appropriate pathway. It's worth noting that the ANDA is based on providing sufficient information to demonstrate that the proposed product is bioequivalent to the Reference Listed Drug (RLD). To do so, the ANDA of a drug-led combination product should also include sufficient information to demonstrate that the device constituent part is compatible for use with the final formulation of the drug constituent part.
At Medidee, now part of Veranex, we have a long track record of providing expertise and technical knowledge to the Medical Device industry. Our Regulatory, Clinical and Quality support for combination products covers all product development stages, to the particular benefit of Pharma companies developing drug-delivery combination products, who may lack a medical device-oriented vision of the regulatory framework.
If this is something you are currently working on, do not hesitate to check our full offering of Combination Products Services, or to reach out directly for expert support.
This article was written by Rima Padovani (PhD, RAC) and Bruno Strappa (MSc).
[UPDATE] Resolution of the German Bundesrat - Urgent need for action in the implementation of the European Medical Devices Regulation (MDR)
The German Länder urge the Federal Government of Germany to take action at the EU level regarding the imminent problems with the implementation of EU Medical Device Regulations.
The Decision 445/22 published on 07.10.2022 summarizes the concerns of the German Länder. See the official publication in German here, and Medidee's tentative translation to English below:
2022-10-09 Summary and tentative translation – Bundesratsbeschluss Oct07-2022
To make sure that your transition to MDR is as quick and smooth as possible, contact Medidee today: www.medidee.com/contacts

[Business Case] Achieving MDR conformity for legacy MDD class I medical software
This Business Case introduces the collaboration between Natural Cycles, a Swedish woman's healthcare company, and Medidee, a global regulatory service provider active in the fields of Medical Devices and IVD products.
When embarking on our journey through evolving regulatory frameworks, Medidee was a stable, informative resource. The experienced team explained detailed guidelines and closely coached us to the point of submission, where the outcome was positive.
Dr Jack Pearson, Medical Affairs Manager at Natural Cycles
Read more about the Quality-Regulatory-Clinical challenges faced by Natural Cycles and how Medidee supported overcoming them:
Business Case – MDR Conformity – Natural Cycles & Medidee
Contact Medidee's experts to discover how we can support you

[Business Case] How the long-term collaboration between Fabrinal and Medidee supports the competitiveness of the Swiss Manufacturer
This Business Case introduces the collaboration between Fabrinal, an SME focused on medical technology applied to studying the eye, and Medidee, a global regulatory service provider active in the fields of Medical Devices and IVD products.
Fabrinal has been supported closely for almost 10 years by Medidee. For an SME of our size, this change is so critical from a timing and economic point of view, that it is crucial to be advised by rational and experienced experts. I am extremely confident about their advice and application of the MDR and can navigate through this challenging change a bit less worried.
Cloé Houriet, CEO of Fabrinal
Read more about the Regulatory challenges faced by Fabrinal and how Medidee supported overcoming them:
Business Case – MDR Transition – Fabrinal & Medidee
Click to contact our experts and get support with your MDR Transition

Artificial Intelligence (AI), General Data Protection Regulation (GDPR) and Cybersecurity: 10 Misconceptions about Medical Device Software
Artificial Intelligence (AI), General Data Protection Regulation (GDPR) and Cybersecurity: 10 Misconceptions about Medical Device Software
Medical Device Software (MDSW) is a growing, fast-evolving industry. However, manufacturers often must face a regulatory framework which does not evolve at the same speed. Regulation for medical devices is restrictive, since it needs to guarantee the safety of users (e.g. Health Care Professionals) and the target population (e.g. patients). Moreover, it has experienced a significant increase in requirements with the approval of the new regulation MDR 2017/745. Manufacturers of MDSW who have never placed a medical device on the market, or who did it under the former Medical Device Directive (93/42/EEC MDD) might have some misconceptions about the process. The purpose of this article is to address some of the most common (and not always right) assumptions and provide useful and truthful information about the process of reaching the conformity assessment under MDR, for successfully placing an MDSW on the market.
Here are 10 common misconceptions about Medical Device Software, and their respective clarification:
1. MDSW is classified as low risk under the MDR 2017/745.
False! On the contrary, only a small portion of MDSW is classified with the lowest risk class (class I) according to the new regulation (MDR2017/745 annex VIII) and related guideline (MDCG 2019-11). To classify a Medical Device Software, two main aspects must be taken into account: 1) the severity of the state of the healthcare situation or patient condition and 2) the significance of the information provided by the software to the healthcare situation related to diagnosis/therapy. After taking these factors into consideration, most MDSW is classified in higher classes, from Class IIa to Class III, which entails increased regulatory requirements.
2. Agile development practice and IEC 62304 requirements cannot co-exist because they rely on fundamentally conflicting principles.
Agile methodologies (i.e. SCRUM) are compatible with the standard for the development of Medical Device Software. Actually, there exists a Technical Information Report providing guidance on the use of Agile practices in the development of medical device software (AAMI TIR45:2012). It is up to the manufacturer to decide the Software Development Lifecycle (SDLC) of the product. However, there are multiple challenges that a manufacturer must face, especially in terms of procedures (alignment with the Quality Management System), validation of tools and documentation.
3. I have developed a Machine Learning model that underwent thorough testing and showed excellent technical performance, so I should be able to access the market in a few months.
All MDSW embedding AI must comply with applicable MDR 2017/745 requirements prior to being placed on the market. This means that the processes and the timing to access the market are not accelerated compared to other medical devices. In addition to the general regulation, there are some relevant specific considerations for the Clinical Evaluation of Medical Device Software as per MDCG 2020-1: For any MDSW (including AI-based MDSW), Clinical Evaluation should demonstrate the valid clinical association/scientific validity, technical performance, and clinical performance. This guidance on clinical evaluation of MDSW provides a framework for the determination of the appropriate level of clinical evidence required for MDSW. The provisions of this guidance document should be taken into consideration from the early stages of software development.
4. I can place my AI-based Software Medical device on the market if I have trained, tested and validated it with datasets coming from open access repositories.
It depends. It is important to verify that sufficient information is available on the origin of the clinical data. Multiple requirements might be fulfilled to ensure the validity of the protocol used to collect the data as well as the compliance of the data collection methods with GDPR: Was the study run according to the Good Clinical Practices and standards? Was GDPR followed? Was the data collection performed by certified professionals? It is also important to adopt good machine learning practices during model training, testing and validation, e.g., that training and testing datasets should be independent. For more information, check these guiding principles for Good Machine Learning Practices.
5. If my MDSW fails to ensure personal data protection, it is not considered as harm.
According to the MDR 2017/745, all parties involved in its application shall respect the confidentiality of information and data obtained. Even if the failure of the software does not result in a lesion or physical injury, disclosure of personal yields infringement penalties according to MDR2017/745 and GDPR Regulation (EU) 2016/679. Therefore, data processing, involving transmission over a network or storage needs to be properly tackled by design strategies (e.g. minimum data collection, pseudo anonymisation) and complemented with ICT techniques (encryption, Secure layers, etc.). To conduct Risk management is a “must”, and any residual risk must be mitigated as much as possible.
6. If my device is not storing data, I do not need to comply with GDPR.
Even if the device does not store data, it still might be, for instance, linked to a website that collects some personal information related to the user or the practitioner. It is important to conduct an analysis of the whole lifecycle of the product and identify which processes need special attention as per the GDPR requirements.
7. If I am working with anonymised data, I do not need to comply with GDPR.
That is true if data is completely anonymised. However, most manufacturers rather work with pseudo-anonymised data, meaning that there is a “key” that can be used to link back the clinical data with the personal information of the patient. In this case, the manufacturer needs to be compliant with GDPR regulations.
8. I can keep the collected data for as long as I want.
Similarly, that depends on whether the collected data is fully anonymised. If that is the case, there are no time restrictions for its storage, but if the data is pseudo-anonymised, there are restrictions. GDPR regulation does not establish specific time windows within which the storage is allowed, instead, it mentions that “personal data must be kept in a form that makes it possible to identify data subjects for no longer than is necessary for the purposes of the processing”.
9. If I use a cloud server, I do not need to worry about cybersecurity because the service provider takes care of it.
Be careful, most cloud servers are not specifically designed to host confidential data or clinical data. When choosing a cloud server for such purposes, it is good to select an ISO 27001 certified provider. That means that the provider has a model for establishing, implementing, operating, monitoring, reviewing, maintaining and improving an information security management system. However, be proactive! Use all relevant information sources (Common Vulnerabilities and Exposures (CVE) for vulnerability monitoring, testing tools such as Trivy, Shodan, OWASP, etc.), and monitor all processes concerning maintenance and infrastructure via health checks.
10. If the Software Medical Device is a standalone software intended to be used in a host, I do not need to take precautions on cybersecurity.
False! MDR 2017/745 requires manufacturers to foresee possible threats caused by misuse of their device and to take actions to prevent it. Besides, MDR also requires reducing as far as possible the risk associated with the possible negative interaction between software and the IT environment the MDSW operates and interacts with. So, it is important to take cybersecurity preventive measures to identify possible threats, vulnerabilities, assets and impacts. A manufacturer needs to consider security in a holistic approach as the nature of assets is diverse: Hardware (including the infrastructure), Software (protection against most common threats such as ransomware, malware, legacy software, Software of Unknown Provenance, etc), Data (Personal Identifiable Information PII, Health Records, Systems configuration, etc), and Users (considering misuse, unauthorised users, protection of sensitive functionalities, etc).
Placing MDSW on the market requires knowledge of a broad variety of topics, including regulation and related guidelines for clinical validation, GDPR, cybersecurity, risk management and quality control.
With an extensive track record working on similar problematics, Medidee can support you with services ranging from training courses and coaching, up to completing the strategy to successfully bring your product to the market.
Contact us today to discuss your project!
This article was written by Dr Nuria Gresa, Dr Stamatia Pagoulatou and Dr Gustavo Hernandez.

[ARTICLE] Why You Need To Review Your Technical Documentation NOW (And 8 Pitfalls to Avoid at all Costs)
According to the Medical Device Survey 2021 by The European Association for Medical devices of Notified Bodies, Medical Device (MD) companies who have applied for MD Regulation (MDR 2017/754) certification remain a minority (only 26.3% had applied at the end of 2021). A lot of applications are yet to be received!
Bottleneck for Technical Documentation (TD) review by Notified Bodies (NB) is a reality
With increased requirements to comply with MDR and a decreased number of Notified Bodies being able to deliver certificates under those regulations (from 50 down to 32 as of August 1st), it gets longer and longer to obtain a certification.
Currently, from the positive outcome of the Completeness Check, it takes between 6 (low-risk devices) and 24 months (high risk)until the issuance of the MDR certificate. Knowing that certificates issued under the MDD/AIMDD remain valid until May 2024 at the latest, waiting too long before submitting your Technical Documentation can result in having no valid certificate to operate in the market.
How to speed up the certification process: be right the first time!
The requirements under MDR are more stringent compared to previous directives. A thorough gap analysis of your Technical Documentation needs to be performed at first to identify the (possibly) missing documents to demonstrate compliance against all General Safety and Performance Requirements (GSPR). Additional device testing might also be required which will significantly delay the moment of application to a NB.
Indeed, with the implementation of the Completeness Check process, at least 50% of the TD submitted are deemed incomplete, and NB request additional information to start the assessment according to the Medical Device Survey 2021. These numbers are confirmed by two other surveys published very recently by the European Commission and MedTech Europe.
Considering these timelines, you cannot afford to wait for the initial feedback from the Notified Body to discover deficiencies in your Technical Documentation – you really need to be right the first time, avoiding common pitfalls.
8 Technical Documentation pitfalls to avoid at all costs
Having performed many gap analyses regarding TD compliance against MDR, as well as having accompanied several companies through MDR transition, Medidee has a clear vision of the main pitfalls throughout this process. Below are typical examples of which companies should be aware before submission. If you would like to learn more about this topic, don't miss the chance to watch our on-demand webinar:

Pitfall 1: Your TD is not well organized
A clear structure throughout the technical documentation is helpful in ensuring that the reviewing body can clearly understand the contents. The MDR now provides, in Annexes II and III, detailed instructions on what is the minimum content of technical documentation, also defining a specific structure for it.
Pitfall 2: No identifiable thread within your TD
Manufacturers shall maintain traceability from the User Requirements Specification (URS) to the Functional Requirements Specification (FRS), risk analysis, clinical evaluation and the GSPR. This is key to demonstrating full compliance to GSPR.
Pitfall 3: Negligence of Post Market Surveillance data
Manufacturers of devices, even those that have been on the market for many years, need to collect or complete a review of existing Post-Market Surveillance (PMS) data, to be able to cover the requirements related to clinical evaluation, as set out by Article 61 of the MDR. For devices that have previously been placed on the market, this includes, but is not limited to the Post Market Clinical Follow Up (PMCF) data, vigilance data, user feedback and complaints.
Pitfall 4: No anticipation of risk class changes for your device
Although this applies to all MDs, it is particularly true for Software as Medical Device (SaMD). Indeed, the classification rule 11 from MDR, and related MDCG guideline MDCG 2019-11, lead to a class upgrade: 80% of the SaMD which did not need a NB before MDR implementation do require one now. This leads to a tremendous change in the company’s organization to obtain a certificate, while also adding to the above-mentioned bottleneck effect.
Pitfall 5: Not enough precision on how GSPR compliance is reached
Manufacturers must provide suitable objective evidence to show that their device satisfies the requirements detailed in Annex I of the MDR GSPRs. Where manufacturers determine that specific GSPRs are not applicable to their device, “an explanation as to why [they] do not apply” must be provided, which is a new requirement in the MDR (Annex II, point 4(a)). Moreover, pointing precisely to the key documents demonstrating compliance to each GSPR will simplify the TD review by the NB as well as help you identify possible gaps within your own TD before submission.
Pitfall 6: No implementation readiness documentation of the UDI traceability system
The Unique Device Identification (UDI) system has a direct impact on the labelling, artwork, and DoC, as manufacturers will need to place a UDI carrier on the label of the device and on all higher levels of packaging, except the shipment packaging (Article 27, point 4). Your quality management system will also need to be aligned to the UDI system implementation. Beware to anticipate any risk linked to the UDI system implementation within your Risk Management file.
Pitfall 7: Absence of specific information relating to device design and history
For all classes of medical devices, manufacturers must now provide, as per Annex II, information in the technical documentation to explain the design stages and procedures that are applied to their device (Annex II, point 3). Under the requirements of the MDD/AIMDD, this was only the case for class III devices. Therefore, depending on the classification of the device, manufacturers may need to update the content of the technical documentation.
Pitfall 8: Person Responsible for Regulatory Compliance (PRRC) not designated early enough within your organization
Article 15 of the MDR clearly stipulates the obligation for medical device manufacturers to have available, within their organization (or permanently and continuously at their disposal for micro and small companies), at least one person, possessing the necessary expertise in the field of medical devices, who is responsible for regulatory compliance. Among other responsibilities, the person or people responsible for regulatory compliance must ensure that the technical documentation is compiled and maintained.
Medidee: Easying your MDR transition with services tailored to your needs
From a simple gap analysis to guide you towards the appropriate tests or data reviews needed, to performing those reviews, building test protocols, writing test reports or consolidating the entire TD, Medidee’s team of experts is ready to support you in this process.
Having accompanied many companies in the transition to MDR, Medidee has gained experience and is able to make this transition as smooth as possible for your organization.
Do not wait any longer! Learn more about our MDR Transition service and contact us today to kick-start your own MDR transition!
This article was written by Dr Lydie Moreau.
[ARTICLE] IVD Manufacturers' New Year's Resolutions: 6 Reasons to Start the Performance Evaluation of your Device Now
Once a device is used for diagnostic purposes on human specimens, the European Union expects, in accordance with Article 5(3) of Regulation (EU) 2017/746 (IVDR), a performance evaluation of the device as a demonstration of compliance with the relevant General Safety and Performance Requirements (GSPRs) and this regardless of the device’s risk class.
As defined within Article 2(44) of the IVDR this means an “assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device”.
The performance evaluation shall be based on a continuous process and follow a defined and methodologically sound procedure, based on an established device-specific plan, i.e., the Performance Evaluation Plan (PEP). The identified performance and safety data, which shall allow to demonstrate compliance with relevant GSPRs related to the device’s performance characteristics, is then consolidated in the Performance Evaluation Report (PER). The PER is a part of the device’s technical documentation and shall be reviewed by the Notified Body during the conformity assessment procedure. De facto, manufacturers of legacy IVD devices will need to invest resources in evaluating the performance of their devices prior to their transition to IVDR.
Here are 6 reasons why manufacturers should not put off this endeavor and start the process now
1. It ensures alignment with the current "state of the art"
In accordance with the requirements of the Regulation (Annex XIII), a PEP must include “a description of the state of the art, including an identification of existing relevant standards, Common Specifications, guidance or best practices documents”. Implementing the PEP as soon as possible will ensure that the device is still aligned with the current state of the art and does not require major design improvements or additional testing according to the latest guidelines. This is especially important because “the performance evaluation of an IVD must consider the benefit-risk ratio in light of the state-of-the-art” and as further stipulated in MDCG 2022-2, “risks for IVDs are often generated from a series of events which may involve several factors such as inadequate design characteristics as well as immature technology”. Therefore, establishing the state of the art is the first step in the preparation of the performance evaluation. As it involves a systematic and methodological approach to the literature review, it requires time and resources.
2. It allows for an early assessment of the level of clinical evidence required
The level of clinical evidence needed to fully demonstrate the performance and safety of the device as claimed by the manufacturer will depend on the characteristics of the device and its intended use. Manufacturers transitioning their devices to IVDR shall assess as soon as possible the level of clinical evidence required. This includes verifying that all performance and safety claims, including those used for marketing purposes, are supported by an adequate level of evidence. Doing so will either allow for timely follow-up actions to collect additional performance data in case some claims are not yet sufficiently supported by clinical evidence, or to reword the device’s intended use taking into consideration the existing clinical evidence.
3. It helps to identify and react to gaps in the analytical performance
Conducting a performance evaluation in advance will allow the manufacturer to identify gaps in the data supporting the analytical performance of the device in a timely manner. If gaps are identified, the manufacturer will be able to generate new evidence in accordance with common specifications, guidelines, or applicable standards, to demonstrate that the IVD in question is capable of reliably, accurately, and consistently detecting the analyte, and thus GSPRSs linked to the analytical features of the device are adequately addressed.
4. It determines if sufficient clinical performance data is available
It is also important to promptly determine whether sufficient clinical performance data is available to support the device's intended use, as well as the claimed indications. The level of clinical evidence should be consistent with the clinical strategy defined in the PEP. As per MDCG 2022-2 “the manufacturer should demonstrate that the IVD has been tested for the intended use(s), target population(s), use condition(s), operating- and use environment(s) and with all the intended user group(s)”. This last aspect is particularly important when it comes to devices intended for point-of-care testing, where manufacturers need to ensure that the clinical data reflects the device’s use environment. Available clinical data may come from the manufacturer’s own clinical performance studies, Post-Market Surveillance (PMS) or peer-reviewed literature. Identifying gaps in advance will allow time to plan and eventually conduct a clinical performance study in compliance with IVDR and ISO 20916:2019, if necessary. It is important to remember that under Article 56(4) of the IVDR, “Clinical performance studies in accordance with Section 2 of Part A of Annex XIII shall be carried out unless it is duly justified to rely on other sources of clinical performance data”.
5. It supports risk identification and management
MDCG 2022-2 (6.2) states that “the risk management system should be carefully aligned with and reflected in the performance evaluation process of the IVD, considering the clinical risks to be addressed as part of the performance evaluation, performance studies, and post-market performance follow-up(s)”. Hence, manufacturers must ensure that all risks identified through the performance evaluation process are adequately addressed within the device’s risk management documents. Timely completion of the performance evaluation will allow the manufacturer sufficient time to address newly identified risks, if necessary.
6. It facilitates robust Post-Market Surveillance and Performance plans
In the event that the time frame is still too short to address all identified performance gaps through appropriate V&V testing or clinical performance studies, the manufacturer will be able to work on a robust PMS and Post-Market Performance Follow-up (PMPF) plan aligned with the performance evaluation outcome. Additionally, in case new PMS and PMPF data were collected after the initial performance evaluation, the PEP and PER should be updated in light of this new data prior to the device’s conformity assessment. As this always requires a certain amount of time for proper implementation, it is best to plan ahead.
In conclusion, the performance evaluation may identify some gaps in the quantity or quality of available clinical evidence or in the completeness of the device’s risk management documentation or PMS/PMPF plan. Depending on the extent and nature of these gaps, it may take time to address them, especially if a clinical performance study is needed, which may require months or years. Additionally, manufacturers should keep in mind that a conformity assessment procedure can take 18-24 months, which means that for legacy IVD devices classified as Class D or C according to IVDR, compliance with GDPR is expected by May 26, 2025 and 2026, respectively.
With this in mind, we advise you to place the performance evaluation of your device as a top priority on your to-do list for 2023. Contact Medidee today to discuss your needs and how Medidee supports you in addressing them: www.medidee.com/contacts
Don't miss any of our upcoming IVD content! Follow Medidee on LinkedIn
This article was written by Dr. Julianne Bobela.
[ARTICLE] Electrical Safety: How changes to IEC 60601 Series trigger additional testing for your device (IEC 60601-1, Edition 3.2)
This article is the first one of a series about IEC 60601. It contains insightful information about IEC 60601-1 Edition 3.2, with a similar structure as the second article.
The latest amendment of IEC 60601-1 for medical electrical equipment - Part 1: General requirements for basic safety and essential performance (IEC 60601-1 Edition 3.2) was published on 2020-08-20 by the International Electrotechnical Commission (IEC). Edition 3.2 notably addresses issues raised by national committees and questions submitted to IEC/SC 62A/Working Group 14 with regards to previous version 3.1.
Since the 30th of May 2022, the United States Food & Drug Administration (FDA) has listed IEC 60601-1 3.2 Edition for Medical Electrical Equipment – Part 1: General requirements for basic safety and essential performance as a recognized standard (FDA recognition number 19-46). IEC 60601-1 3.2 Edition will replace the current listed IEC 60601-1 3.1 Edition (FDA recognition number 19-4) by the 17th of December 2023. Until this date, FDA will accept declarations of conformity referring to IEC 60601-1 3.1 Edition, in support of premarket submissions.
To date, no announcement relative to an official transition period for the applicability of Edition 3.2 or to its harmonization under the Medical Device Directive (93/42/EEC) or Medical Device Regulation (EU 2017/745) has been issued in the official journal of the European Union. As per the list of recognized consensus standards of the Food & Drug Administration (FDA), ES60601-1:2005/(R)2012 and A1:2012, C1:2009/(R)2012 and A2:2010/(R)2012 (Consolidated Text) are currently listed.
In the absence of any official communication within the European Union on the matter, it is anticipated that manufacturers will be granted a transition period of 3 to 4 years after the date of publication, before they will be expected to demonstrate compliance with the applicable new requirements introduced by the amended standard.
Medidee has observed that manufacturers of electrically driven active medical devices are commonly unaware of the latest revision of this standard and consequently of the new requirements it entails. This article aims to raise awareness of this amendment of IEC 60601-1, to summarize the main changes compared to the previous version of the standard, and to illustrate through a few examples how these changes may affect manufacturers of medical electrical equipment and systems.
What are main the differences between IEC 60601-1 Edition 3.1 and Edition 3.2?
The main clauses affected in IEC 60601-1 Edition 3.2 are:
Clause 2 normative references
Clause 3 terminology and definitions
Clause 7 identification and marking
Clause 8 protection against electrical hazards
Clause 11 protection against excessive temperatures and other hazards
Clause 13 hazardous situations and fault conditions
Clause 14 programmable electrical medical systems (PEMS)
Importantly, the normative references of the standard have also been updated in Edition 3.2. The changes to referenced standards are shown in bold text below:
IEC 60065:2001+AMD1:2005+AMD2:2010 Audio, video and similar electronic apparatus
IEC 60068-2-2:2007 Environmental testing
IEC 60227-1:2007 Insulated cables
IEC 60245-1:2003+AMD1:2007 Rubber insulated cables – general requirements
IEC 60335-1:2010 Household and similar appliances
IEC 60417 Graphical symbols for use on equipment
IEC 60601-1-2:2014+AMD1:2020 Electromagnetic disturbances – requirements and tests
IEC 60601-1-3:2008+AMD1:2013 Radiation protection in diagnostic X-ray equipment
IEC 60601-1-6:2010+AMD1:2013+AMD2:2020 Usability
IEC 60601-1-8:2006+AMD1:2012+AMD2:2020 Alarm systems in medical electrical equipment
IEC 60664-1:2007 Insulation coordination
IEC 60730-1:2014 Automatic electrical controls for household and similar use
IEC 60747-5-5:2007 Semiconductor devices – optoelectronic devices - photocouplers
IEC 60825-1:2014 Safety of laser products
IEC 60065:2001+AMD1:2005+AMD2:2010 Audio, video and similar electronic apparatus IEC 60068-2-2:2007 Environmental testing
IEC 60227-1:2007 Insulated cables
IEC 60245-1:2003+AMD1:2007 Rubber insulated cables – general requirements
IEC 60335-1:2010 Household and similar appliances
IEC 60417 Graphical symbols for use on equipment
IEC 60601-1-2:2014+AMD1:2020 Electromagnetic disturbances – requirements and tests IEC 60601-1-3:2008+AMD1:2013 Radiation protection in diagnostic X-ray equipment
IEC 60601-1-6:2010+AMD1:2013+AMD2:2020 Usability
IEC 60601-1-8:2006+AMD1:2012+AMD2:2020 Alarm systems in medical electrical equipment
IEC 60664-1:2007 Insulation coordination
IEC 60730-1:2014 Automatic electrical controls for household and similar use
IEC 60747-5-5:2007 Semiconductor devices – optoelectronic devices - photocouplers
IEC 60825-1:2014 Safety of laser products
IEC 60851-3:2009 Winding wires – test methods mechanical properties
IEC 60851-5:2008 Winding wires – test methods electrical properties
IEC 60950-1:2005+AMD1:2009+AMD2:2013 Information technology equipment
IEC 61058-1:2000+AMD1:2001+AMD2:2007 Switches for appliances
IEC 62133 Secondary cells and batteries
IEC 62133-2 Secondary cells and batteries (Lithium systems)
IEC 62304:2006+AMD1:2015 Medical device software – software life cycle processes
IEC 62368-1:2018 Audio/ video, information, and communication technology equipment
ISO 7010:2019 Graphical symbols
ISO 11135-1:2007 Sterilization of health care products – Ethylene oxide
ISO 11137-1:2006 Sterilization of health care products – requirements for validation
ISO 13857:2008 Safety of machinery
ISO 14971:2019 Medical devices – application of risk management
ISO 15223-1:2016 Medical devices – symbols used with medical device labels
ISO 17665-1:2006 Sterilization of health care products – requirements for validation (moist heat)
ISO 80000-1:2009 Quantities and units
What do these changes mean in practice?
Due to the technological progress and resulting technical changes, the standards to ensure a safe and effective medical device needs to develop too (state of the art). To comply with IEC 60601-1 3.2 edition the latest editions of applicable process, particular, collateral, and component standards need to be considered by the medical device applicant.
Medical device manufacturers are advised to take advantage of the transition period to perform a gap analysis of their technical documentation (TD) against Edition 3.2 and update their documentation accordingly. Once potential gaps have been cleared, a new test report according to IEC 60601-1:2005+AMD1:2012+AMD2:2020 Edition 3.2 can be requested from an external ISO/IEC 17025 accredited testing laboratory. If the current IEC 60601-1 test report uses CB-Scheme as test scheme, it should be kept in mind that according to IEC Operational Document OD-2037 clause 3.1, a new Certification Body (CB) test certificate shall be issued with a new CB test certificate number. Thanks to the fact that now the TD will have been revised to comply with technical state of the art, a smooth certification process will be assured.
Let's focus on the changes to IEC 60601-1 clause 8 - protection against electrical hazards and clause 14 - programmable electrical medical systems. One noticeable change to clause 8 concerns the introduction of the requirement to measure the voltage of all accessible conductive parts of the Signal Input/ Signal Output (SIP/SOP) or power output connectors to earth for medical electrical equipment which is equipped with SIP/SOP connectors or separate power supply output connectors. Depending on whether the voltage is greater than 60Vdc or 42,4Vac - touch current measurement according to clause 8.7.3 will need to be conducted. For measuring the voltage of the SIP/SOP, please refer to the below-mentioned electrical schematic.
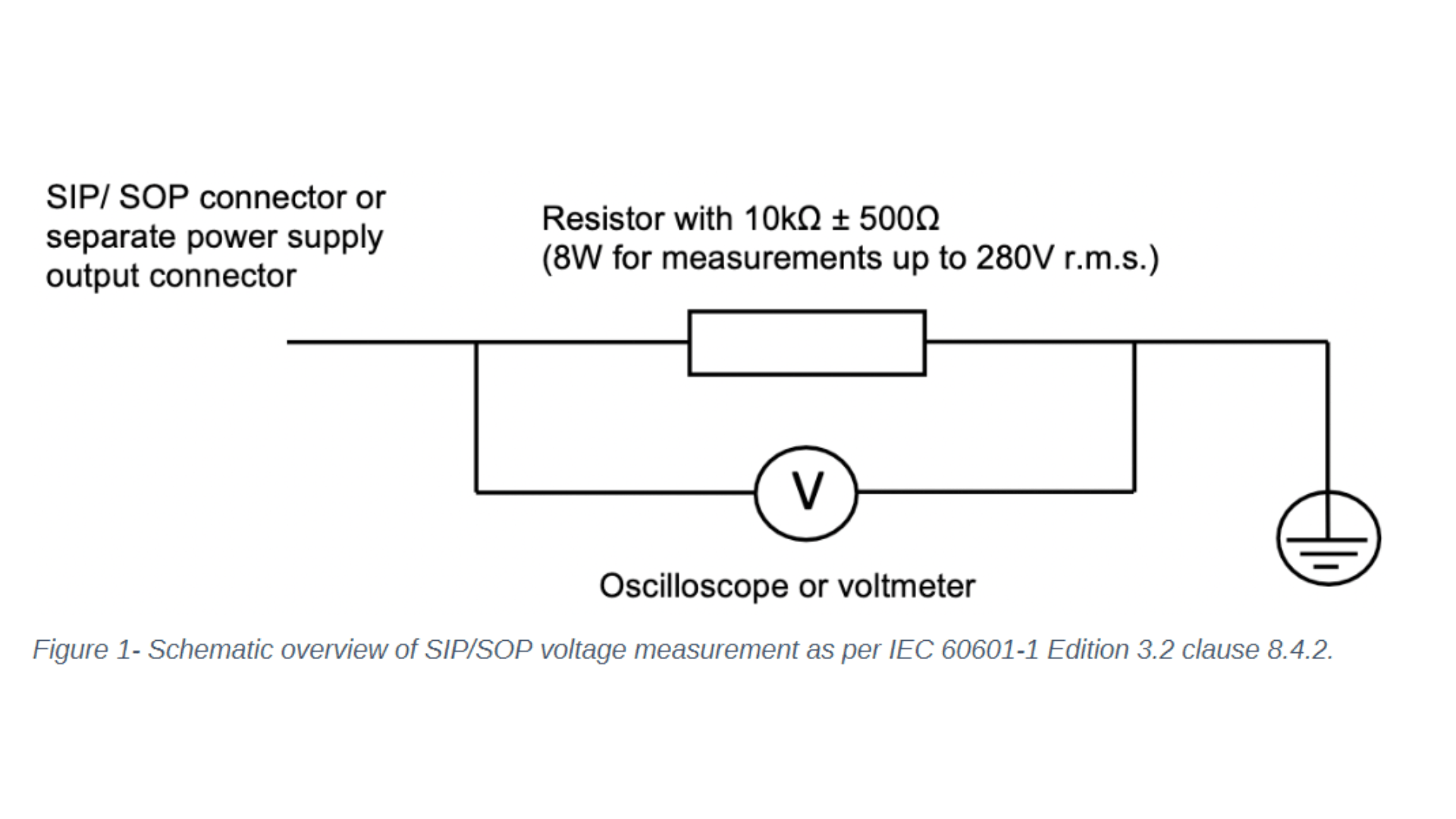
Another noteworthy update of IEC 60601-1 Edition 3.2 affecting clause 8 is the addition of the normative reference to the standard IEC 60747-5-5:2007 Semiconductor devices – optoelectronic devices – photocouplers. Optocouplers which comply with IEC 60747-5-5:2007 or a later edition are considered equivalent to the requirements of 8.8.2 for distances through solid insulation and 8.9.3 for spaces filled by insulation compound. If your medical electrical device or system uses an optocoupler as a solid insulation forming a means of operator protection, the following verification measures apply and need to be conducted:
Air clearance at the outside of the opto-coupler
Creepage distance at the outside of the opto-coupler
Dielectric strength across the opto-coupler
Moreover, Clause 14 Programmable Electrical Medical Systems (PEMS) introduced a direct reference to IEC 62304:2006+AMD1:2015 Medical device software – software life cycle processes. Please keep in mind that the requirements in subclauses 14.2 to 14.12 shall apply to PEMS if the medical electrical equipment uses Programmable Electronics Subsystem (PESS) to provide functionality necessary for basic safety or essential performance or if the application of risk management as described in 4.2 shows that the failure of any PESS leads to an unacceptable risk. Medical device manufacturers are required to evaluate if clause 14 of IEC 60601-1 - and therefore specific clauses of IEC 62304 - are applicable.
How can Medidee support your company?
Our team of electrical safety specialists at Medidee assist you in identifying all areas requiring updates to meet the new requirements introduced by IEC 60601-1 Edition 3.2 through a detailed review of the technical documentation of your medical electrical device or system, as well as provide support in the implementation of applicable new IEC 60601-1 Edition 3.2 requirements. For instance, Medidee reviews and updates the list of critical components and insulation diagram, and process referenced standard-related changes incurred by the update of referenced standards ISO 14971:2019, IEC 62304:2006+AMD1:2015, and IEC 60601-1-6:2010+AMD1:2013+AMD2:2020/ IEC 62366-1:2015+AMD1:2020.
In addition, Medidee supports you in communicating with external accredited testing laboratories and notified bodies, to ensure a smooth certification process of the medical electrical equipment or system.
Contact Medidee today to discuss your needs and how Medidee supports you in addressing them: www.medidee.com/contacts
This article was written by Stefan Staltmayr.