[ARTICLE] Combination Products: Similarities and Differences of EU and US Regulations
The success of novel combination products is much dependent on the regulatory intelligence and foresight to define the best strategy to place and maintain them on the market, through a deep understanding of how drugs, devices and their combination are regulated in the different target markets.
This article aims to provide general guidelines for combination product manufacturers who may not be familiar with the EU and US regulations, or know one of these markets well and aim to access the other.
Terminology and definitions
The term “combination product”, commonly used to identify the category of medical products made of a combination of drug, device, and/or biologic, comes from the FDA definition given in 21 CFR 3.2 (e).
In the EU, instead, the wording “integral product” is used to refer to devices incorporating a medicinal substance (Article 1 §8), and devices intended to administer a medicinal product (Article 1 §9), as defined in the Medical Device Regulation (EU) 2017/745 (MDR). Of note, the European Medicines Agency (EMA) has published a Q&A guidance document on this topic and the term “Drug Device Combination” (DDC) is also used.
In 21 CFR 3.2 (e), the FDA defines the following types of combination products:
- single-entity combination product in which its constituent parts are physically, chemically, or otherwise combined or mixed and produced, as a single entity;
- co-packaged combination product in which its constituent parts are packaged together in a single package or as a unit;
- cross-labeled combination product in which its constituent parts are packaged separately and, according to the investigational plan or proposed labeling, are intended for use together to achieve the intended use, indication, or effect.
According to the MDR, the same products are categorized differently, as follows:
- devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device;
- devices incorporating a medicinal substance as an integral part which has an action principal to that of the device;
- devices intended to administer a medicinal product, placed on the market as a single integral product (i.e. placed on the market in such a way that they form a single integral product which is intended exclusively for use in the given combination and which is not reusable);
- devices intended to administer a medicinal product, placed on the market as a “non-integral product”.
To provide some examples, a pre-filled syringe is a single-entity combination product in the US, and “a device intended to administer a medicinal product as a single integral product” in the EU.
A drug-eluting stent is a single-entity combination product in the US, and “a device incorporating a medicinal substance as an integral part which has an action ancillary to that of the device” in the EU.
In the US, insulin cartridges to be used with a reusable pen injector can be a co-packaged combination product, if the insulin cartridges are provided in the same package with the reusable pen, or a cross-labeled combination product, if provided separately. In the EU, the reusable pen injector is “a device intended to administer a medicinal product as a non-integral product”.
Mode of Action to define how the product is regulated
Interestingly, the term “mode of action” is used both in the US and in the EU, but its definition is provided only in 21 CFR 3.2(k), which states that the “mode of action is the means by which a product achieves an intended therapeutic effect or action”.
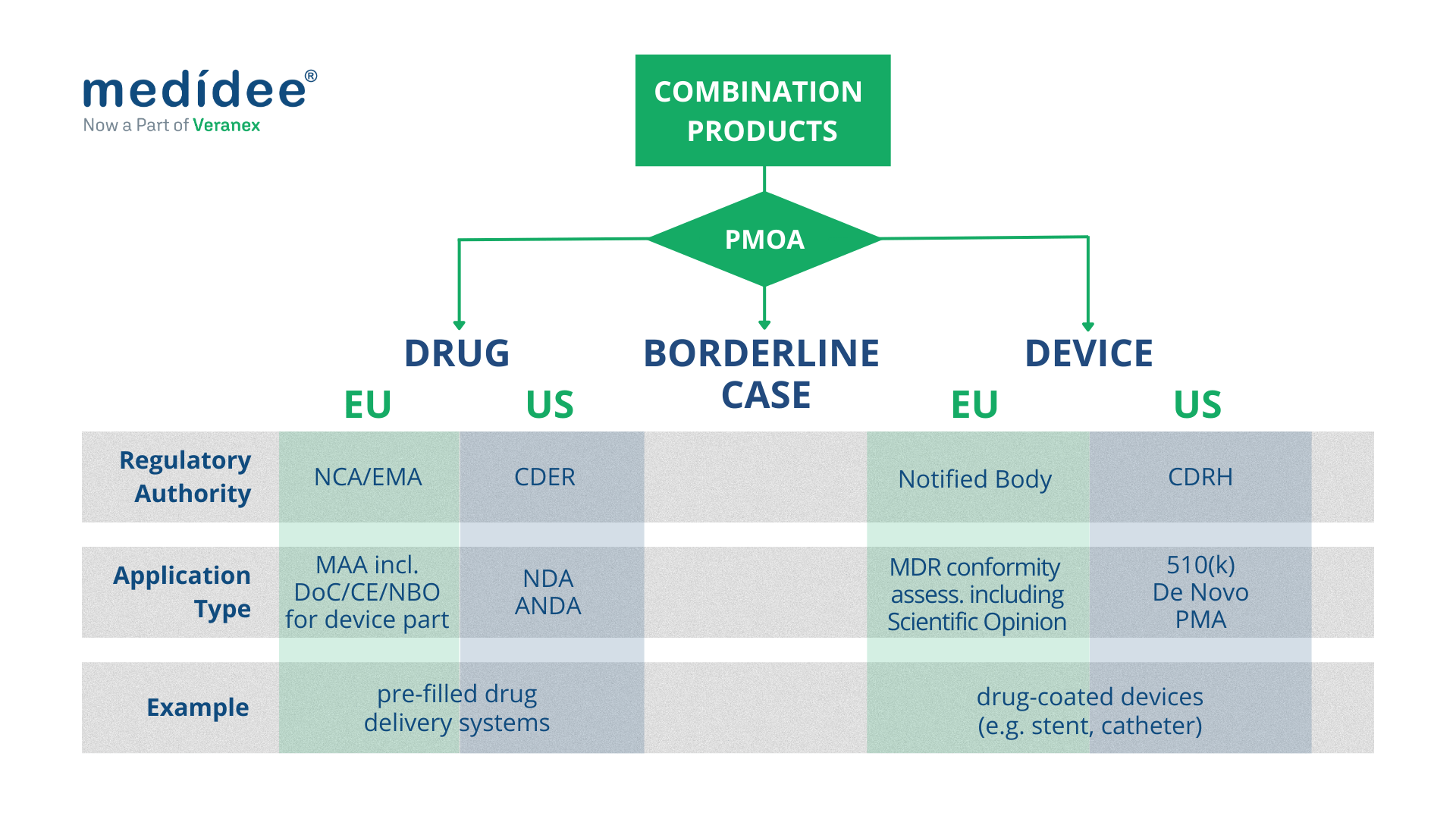
Both in the US and in the EU, combination products are recognized to have more than one identifiable mode of action. Among the multiple modes of action, FDA defines as Primary Mode of Action (PMOA) “the single mode of action of a combination product that provides the most important therapeutic action of the combination product. The most important therapeutic action is the mode of action expected to make the greatest contribution to the overall intended therapeutic effects of the combination product.” (21 CFR 3.2(m)).
The MDR does not provide a definition, but rather distinguishes between “principal” and “ancillary” action of the medicinal substance to that of the device.
Finally, the “primary” or “principal” mode of action is the key criterion used in both US and EU’s regulatory frameworks to define how the product is regulated and by which Regulatory Authority.
Combination Products regulated as medical devices
Both in the EU and in the US, combination products with a device PMOA, also called device-led combination products, are regulated as medical devices.
European Union
In the EU, this type of combination product includes “devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device”, and “devices intended to administer a medicinal product, placed on the market as a non-integral product”. They undergo conformity assessment procedures as any other medical device, as defined in Article 52 and set out in Annex IX to XI of the MDR.
In terms of classification, “devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device” are class III medical devices, according to Rule 14, as defined in Annex VIII of the MDR.
Instead, “devices intended to administer a medicinal product, placed on the market as a non-integral product” are classified differently depending on their intended purpose. Of note, the MDR up-classified some of these products: for example, inhalers were regulated under the Medical Device Directive 93/42/EEC (MDD) as class I devices, whereas they are now class IIa or class IIb devices under the MDR.
Finally, as part of the conformity assessment procedure, “devices incorporating a medicinal substance as an integral part which has an action ancillary to that of the device” require consultation by the Notified Body of the relevant Medicinal Products Authority (National Competent Authority or EMA), to seek their Scientific Opinion. This consultation process should be taken into account when planning the CE marking of such products or when planning a significant change to an already CE-marked product, as it greatly impacts the timelines for completion of the conformity assessment procedure.
United States
In the US, as a general rule, FDA requires a single application to streamline regulatory interactions with the Agency and the marketing application type generally coincides with the PMOA of the combination product. Hence for a device-led combination product, a single application should be submitted to the Center for Devices and Radiological Health (CDRH) which acts as “lead center”.
A Premarket Approval (PMA) is required for class III device-led combination products; a De Novo Classification Request applies for novel device-led combination products for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use (class I and class II); a Premarket Notification (510(k)) applies when the substantial equivalence of the device-led combination product can be demonstrated against a predicate product.
Of note that, as part of the single application, sufficient information should be provided on the drug constituent part and may include nonclinical pharmacology, toxicology and clinical pharmacology (including pharmacokinetic) data and chemistry, manufacturing, and controls (CMC) information.

Combination Products regulated as drugs
Both in the EU and in the US, combination products with a drug PMOA, also called drug-led combination products, are regulated as drugs.
European Union
In the EU, “Devices incorporating a medicinal substance as an integral part which has an action principal to that of the device” and “devices intended to administer a medicinal product, placed on the market as a single integral product” are regulated as medicinal products under Directive 2001/83/EC or Regulation (EC) No 726/2004, as applicable.
In this context, Article 117 of the MDR introduces an amendment to Directive 2001/83/EC, requiring that the Marketing Authorisation Application (MAA) includes the results of the assessment of the conformity of the device part with the relevant General Safety and Performance Requirements (GSPRs) set out in Annex I of the MDR. This translates into two cases:
- if the device part classifies as a class I medical device, then the evidence of conformity to the relevant GSPRs is provided as part of the manufacturer’s EU Declaration of Conformity;
- if the device part classifies in any other way (i.e. class Im, class Is, class Ir, class IIa, class IIb, and class III), then the evidence of conformity to the relevant GSPRs is provided either as the CE certificate issued by a Notified Body (if the device is CE marked), or as the so-called Notified Body Opinion. In the latter case, careful planning of the submission of the Technical Documentation of the device part to the Notified Body must be done to ensure sufficient time for review by the Notified Body and obtain the Notified Body Opinion, to be included as part of the MAA.
United States
In the US, for a drug-led combination product, a single application should be submitted to the Center for Drug Evaluation and Research (CDER) which, acts as “lead center”.
For a new drug-led combination product, the single application is a New Drug Application (NDA); for drug-led combination products that are the generic version of already-approved drug-led combination products, the Abbreviated New Drug Application (ANDA) is, in general, the most appropriate pathway. It’s worth noting that the ANDA is based on providing sufficient information to demonstrate that the proposed product is bioequivalent to the Reference Listed Drug (RLD). To do so, the ANDA of a drug-led combination product should also include sufficient information to demonstrate that the device constituent part is compatible for use with the final formulation of the drug constituent part.
At Medidee, now part of Veranex, we have a long track record of providing expertise and technical knowledge to the Medical Device industry. Our Regulatory, Clinical and Quality support for combination products covers all product development stages, to the particular benefit of Pharma companies developing drug-delivery combination products, who may lack a medical device-oriented vision of the regulatory framework.
If this is something you are currently working on, do not hesitate to check our full offering of Combination Products Services, or to reach out directly for expert support.
This article was written by Rima Padovani (PhD, RAC) and Bruno Strappa (MSc).