[ARTICLE] Electrical Safety: How changes to IEC 60601 Series trigger additional testing for your device (IEC 60601-1, Edition 3.2)
This article is the first one of a series about IEC 60601. It contains insightful information about IEC 60601-1 Edition 3.2, with a similar structure as the second article.
The latest amendment of IEC 60601-1 for medical electrical equipment - Part 1: General requirements for basic safety and essential performance (IEC 60601-1 Edition 3.2) was published on 2020-08-20 by the International Electrotechnical Commission (IEC). Edition 3.2 notably addresses issues raised by national committees and questions submitted to IEC/SC 62A/Working Group 14 with regards to previous version 3.1.
Since the 30th of May 2022, the United States Food & Drug Administration (FDA) has listed IEC 60601-1 3.2 Edition for Medical Electrical Equipment – Part 1: General requirements for basic safety and essential performance as a recognized standard (FDA recognition number 19-46). IEC 60601-1 3.2 Edition will replace the current listed IEC 60601-1 3.1 Edition (FDA recognition number 19-4) by the 17th of December 2023. Until this date, FDA will accept declarations of conformity referring to IEC 60601-1 3.1 Edition, in support of premarket submissions.
To date, no announcement relative to an official transition period for the applicability of Edition 3.2 or to its harmonization under the Medical Device Directive (93/42/EEC) or Medical Device Regulation (EU 2017/745) has been issued in the official journal of the European Union. As per the list of recognized consensus standards of the Food & Drug Administration (FDA), ES60601-1:2005/(R)2012 and A1:2012, C1:2009/(R)2012 and A2:2010/(R)2012 (Consolidated Text) are currently listed.
In the absence of any official communication within the European Union on the matter, it is anticipated that manufacturers will be granted a transition period of 3 to 4 years after the date of publication, before they will be expected to demonstrate compliance with the applicable new requirements introduced by the amended standard.
Medidee has observed that manufacturers of electrically driven active medical devices are commonly unaware of the latest revision of this standard and consequently of the new requirements it entails. This article aims to raise awareness of this amendment of IEC 60601-1, to summarize the main changes compared to the previous version of the standard, and to illustrate through a few examples how these changes may affect manufacturers of medical electrical equipment and systems.
What are main the differences between IEC 60601-1 Edition 3.1 and Edition 3.2?
The main clauses affected in IEC 60601-1 Edition 3.2 are:
Clause 2 normative references
Clause 3 terminology and definitions
Clause 7 identification and marking
Clause 8 protection against electrical hazards
Clause 11 protection against excessive temperatures and other hazards
Clause 13 hazardous situations and fault conditions
Clause 14 programmable electrical medical systems (PEMS)
Importantly, the normative references of the standard have also been updated in Edition 3.2. The changes to referenced standards are shown in bold text below:
IEC 60065:2001+AMD1:2005+AMD2:2010 Audio, video and similar electronic apparatus
IEC 60068-2-2:2007 Environmental testing
IEC 60227-1:2007 Insulated cables
IEC 60245-1:2003+AMD1:2007 Rubber insulated cables – general requirements
IEC 60335-1:2010 Household and similar appliances
IEC 60417 Graphical symbols for use on equipment
IEC 60601-1-2:2014+AMD1:2020 Electromagnetic disturbances – requirements and tests
IEC 60601-1-3:2008+AMD1:2013 Radiation protection in diagnostic X-ray equipment
IEC 60601-1-6:2010+AMD1:2013+AMD2:2020 Usability
IEC 60601-1-8:2006+AMD1:2012+AMD2:2020 Alarm systems in medical electrical equipment
IEC 60664-1:2007 Insulation coordination
IEC 60730-1:2014 Automatic electrical controls for household and similar use
IEC 60747-5-5:2007 Semiconductor devices – optoelectronic devices - photocouplers
IEC 60825-1:2014 Safety of laser products
IEC 60065:2001+AMD1:2005+AMD2:2010 Audio, video and similar electronic apparatus IEC 60068-2-2:2007 Environmental testing
IEC 60227-1:2007 Insulated cables
IEC 60245-1:2003+AMD1:2007 Rubber insulated cables – general requirements
IEC 60335-1:2010 Household and similar appliances
IEC 60417 Graphical symbols for use on equipment
IEC 60601-1-2:2014+AMD1:2020 Electromagnetic disturbances – requirements and tests IEC 60601-1-3:2008+AMD1:2013 Radiation protection in diagnostic X-ray equipment
IEC 60601-1-6:2010+AMD1:2013+AMD2:2020 Usability
IEC 60601-1-8:2006+AMD1:2012+AMD2:2020 Alarm systems in medical electrical equipment
IEC 60664-1:2007 Insulation coordination
IEC 60730-1:2014 Automatic electrical controls for household and similar use
IEC 60747-5-5:2007 Semiconductor devices – optoelectronic devices - photocouplers
IEC 60825-1:2014 Safety of laser products
IEC 60851-3:2009 Winding wires – test methods mechanical properties
IEC 60851-5:2008 Winding wires – test methods electrical properties
IEC 60950-1:2005+AMD1:2009+AMD2:2013 Information technology equipment
IEC 61058-1:2000+AMD1:2001+AMD2:2007 Switches for appliances
IEC 62133 Secondary cells and batteries
IEC 62133-2 Secondary cells and batteries (Lithium systems)
IEC 62304:2006+AMD1:2015 Medical device software – software life cycle processes
IEC 62368-1:2018 Audio/ video, information, and communication technology equipment
ISO 7010:2019 Graphical symbols
ISO 11135-1:2007 Sterilization of health care products – Ethylene oxide
ISO 11137-1:2006 Sterilization of health care products – requirements for validation
ISO 13857:2008 Safety of machinery
ISO 14971:2019 Medical devices – application of risk management
ISO 15223-1:2016 Medical devices – symbols used with medical device labels
ISO 17665-1:2006 Sterilization of health care products – requirements for validation (moist heat)
ISO 80000-1:2009 Quantities and units
What do these changes mean in practice?
Due to the technological progress and resulting technical changes, the standards to ensure a safe and effective medical device needs to develop too (state of the art). To comply with IEC 60601-1 3.2 edition the latest editions of applicable process, particular, collateral, and component standards need to be considered by the medical device applicant.
Medical device manufacturers are advised to take advantage of the transition period to perform a gap analysis of their technical documentation (TD) against Edition 3.2 and update their documentation accordingly. Once potential gaps have been cleared, a new test report according to IEC 60601-1:2005+AMD1:2012+AMD2:2020 Edition 3.2 can be requested from an external ISO/IEC 17025 accredited testing laboratory. If the current IEC 60601-1 test report uses CB-Scheme as test scheme, it should be kept in mind that according to IEC Operational Document OD-2037 clause 3.1, a new Certification Body (CB) test certificate shall be issued with a new CB test certificate number. Thanks to the fact that now the TD will have been revised to comply with technical state of the art, a smooth certification process will be assured.
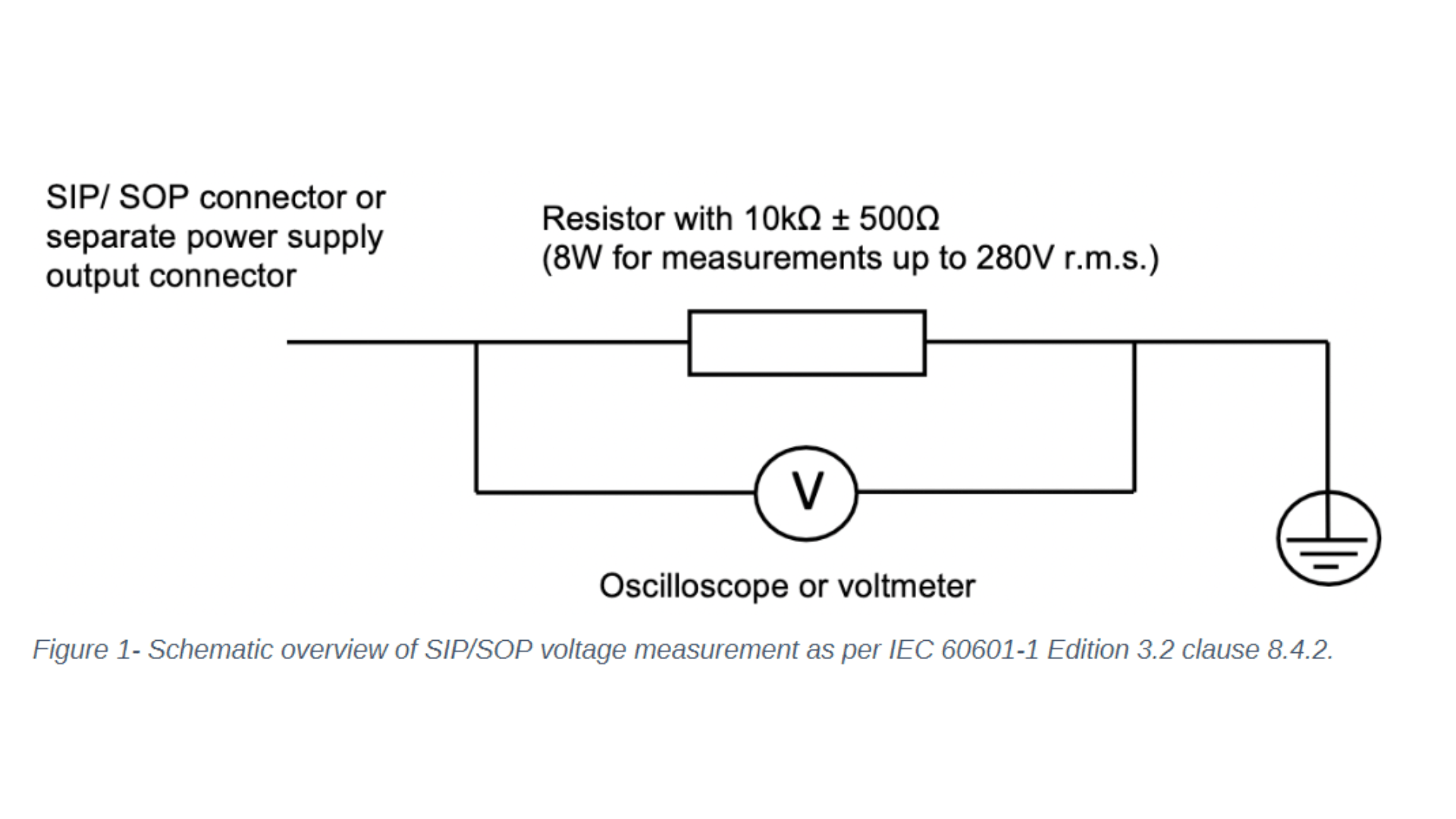
Let's focus on the changes to IEC 60601-1 clause 8 - protection against electrical hazards and clause 14 - programmable electrical medical systems. One noticeable change to clause 8 concerns the introduction of the requirement to measure the voltage of all accessible conductive parts of the Signal Input/ Signal Output (SIP/SOP) or power output connectors to earth for medical electrical equipment which is equipped with SIP/SOP connectors or separate power supply output connectors. Depending on whether the voltage is greater than 60Vdc or 42,4Vac - touch current measurement according to clause 8.7.3 will need to be conducted. For measuring the voltage of the SIP/SOP, please refer to the below-mentioned electrical schematic.
Another noteworthy update of IEC 60601-1 Edition 3.2 affecting clause 8 is the addition of the normative reference to the standard IEC 60747-5-5:2007 Semiconductor devices – optoelectronic devices – photocouplers. Optocouplers which comply with IEC 60747-5-5:2007 or a later edition are considered equivalent to the requirements of 8.8.2 for distances through solid insulation and 8.9.3 for spaces filled by insulation compound. If your medical electrical device or system uses an optocoupler as a solid insulation forming a means of operator protection, the following verification measures apply and need to be conducted:
Air clearance at the outside of the opto-coupler
Creepage distance at the outside of the opto-coupler
Dielectric strength across the opto-coupler
Moreover, Clause 14 Programmable Electrical Medical Systems (PEMS) introduced a direct reference to IEC 62304:2006+AMD1:2015 Medical device software – software life cycle processes. Please keep in mind that the requirements in subclauses 14.2 to 14.12 shall apply to PEMS if the medical electrical equipment uses Programmable Electronics Subsystem (PESS) to provide functionality necessary for basic safety or essential performance or if the application of risk management as described in 4.2 shows that the failure of any PESS leads to an unacceptable risk. Medical device manufacturers are required to evaluate if clause 14 of IEC 60601-1 - and therefore specific clauses of IEC 62304 - are applicable.
How can Medidee support your company?
Our team of electrical safety specialists at Medidee assist you in identifying all areas requiring updates to meet the new requirements introduced by IEC 60601-1 Edition 3.2 through a detailed review of the technical documentation of your medical electrical device or system, as well as provide support in the implementation of applicable new IEC 60601-1 Edition 3.2 requirements. For instance, Medidee reviews and updates the list of critical components and insulation diagram, and process referenced standard-related changes incurred by the update of referenced standards ISO 14971:2019, IEC 62304:2006+AMD1:2015, and IEC 60601-1-6:2010+AMD1:2013+AMD2:2020/ IEC 62366-1:2015+AMD1:2020.
In addition, Medidee supports you in communicating with external accredited testing laboratories and notified bodies, to ensure a smooth certification process of the medical electrical equipment or system.
Contact Medidee today to discuss your needs and how Medidee supports you in addressing them: www.medidee.com/contacts
This article was written by Stefan Staltmayr.

[ARTICLE] Electromagnetic Compatibility: How changes to IEC 60601 Series trigger additional testing for your device (IEC 60601-1-2, Edition 4.1)
This article is the second one of a series about IEC 60601. It contains insightful information about amendment 1 of IEC 60601-1-2, following a similar structure as the first article.
The latest amendment 1 of IEC 60601-1-2 for Medical electrical equipment - Part 1-2: General requirements for basic safety and essential performance - Collateral Standard: Electromagnetic disturbances - Requirements and tests (IEC 60601-1-2 Edition 4.1) was published on 2020-09-01 by the International Electrotechnical Commission (IEC). Edition 4.1 notably addresses issues raised by national committees and questions submitted to IEC/SC 62A with regard to previous version 4.0.
Since the 21st of December 2020, the United States Food & Drug Administration (FDA) has listed IEC 60601-1-2 4.1 Edition Medical electrical equipment - Part 1-2: General requirements for basic safety and essential performance - Collateral Standard: Electromagnetic disturbances - Requirements and tests as a recognized standard (FDA recognition number 19-36). IEC 60601-1-2 4.1 Edition will replace the current listed IEC 60601-1-2 4th Edition (FDA recognition number 19-8) by the 17th of December 2023. Until this date, FDA will accept declarations of conformity referring to IEC 60601-1-2 4th Edition, in support of the premarket submission.
To date, no announcement relative to an official transition period for the applicability of Edition 4.1 or to its harmonization under the Medical Device Directive (93/42/EEC) or Medical Device Regulation (EU 2017/745) has been issued in the official journal of the European Union.
In the absence of any official communication within the European Union on the matter, it is anticipated that manufacturers will be granted a transition period of 3 to 4 years after the date of publication before they will be expected to demonstrate compliance with the applicable new requirements introduced by the amended standard.
WHAT ARE THE MAIN DIFFERENCES BETWEEN CURRENT IEC 60601-1-2 EDITION 4.0 AND EDITION 4.1?
Clauses affected in IEC 60601-1-2 Edition 4.1 compared to 4.0 Edition are:
Clause 2 Normative references
Clause 3 Terms and definitions
Clause 4.3.3 Power input voltages and frequencies Table 1 – Power input voltages and frequencies during the tests
Clause 7.1.12 Permanently installed large ME equipment and large ME systems
Clause 7.3 Emissions requirements summary Table 2 – Emission limits per environment
Clause 8.1 Electromagnetic immunity requirements for ME equipment and ME systems - general
Clause 8.6 Permanently installed large ME equipment and large ME systems
Clause 8.9 Immunity test levels
Clause 8.11 Immunity to proximity magnetic fields in the frequency range 9kHz to 13,56 MHz
Figure 3 – Examples of locations within EM environments
Table 4 – Enclosure Port
Table 5 – Input a.c. power port (1 of 2 and 2 of 2)
Table 8 – SIP/SOP port
Table 9 – Test specifications for enclosure port immunity to RF wireless communications equipment
Table 10 – minimum test report contents (2 of 2)
Table 11 – Test specifications for enclosure port immunity to proximity magnetic fields
Table A.3 – Test specifications for enclosure port immunity to RF wireless communications equipment
Annex A.1 Safety and performance
Subclause 4.2 – Non-ME equipment used in an ME system
Subclause 4.3.3 – Power input voltages and frequencies
Subclause 5.2.2.1 a), Compliance with each emission and immunity standard
Subclause 7.1.12 – Permanently installed large ME equipment and Large ME systems
Subclause 8.6 – Permanently installed large ME equipment and large ME systems
Subclause 8.9 – Immunity test levels b) Environments
Subclause 8.9 – Immunity test levels c) Immunity test level determination
Subclause 8.10 – Immunity to proximity fields from RF wireless communications equipment
Subclause 8.11 – Immunity to proximity magnetic fields in the frequency range 9 kHz to 13,56 MHz
Annex F Guidance on the application of risk management with regard to electromagnetic disturbances in this collateral standard
Figure F.1 – Risk management flow in IEC 60601-1-2
Table G.1 Recommended minimum test plan contents (2 of 2)
Importantly, the normative references of the standard have also been updated in Edition 4.1. The changes to referenced standards are shown in bold text below:
IEC 60601-1:2005+AMD1:2012+AMD2:2020
IEC 60601-1-8:2006+AMD1:2012+AMD2:2020
IEC 60601-1-11:2015+AMD1:2020
IEC 60601-1-12:2014+AMD1:2020
IEC 61000-4-5:2014+AMD1:2017
IEC 61000-4-11:2004+AMD1:2017
IEC 61000-4-39:2017
CISPR 11:2015+AMD1:2016+AMD2:2019
CISPR 14-1:2016
CISPR 16-1-2:2014+AMD1:2017
CISPR 32:2015
ISO 14971:2019
WHAT DO THESE CHANGES MEAN IN PRACTICE?
Due to technological progress and resulting technical changes, the standards to ensure a safe and effective medical device need to develop too (state of the art). To comply with IEC 60601-1-2 4.1 edition, the latest editions of applicable process, particular, collateral, and component standards need to be considered by the medical device applicant.
Medical device manufacturers are advised to take advantage of the transition period to perform a gap analysis of their technical documentation (TD) against Edition 4.1 and update their documentation accordingly. Once potential gaps have been cleared, a new test report according to IEC 60601-1-2:2014+AMD1:2020 Edition 4.1 can be requested from an external ISO/IEC 17025 accredited testing laboratory. If the current IEC 60601-1-2 test report uses CB-Scheme as test scheme, it should be kept in mind that according to IEC Operational Document OD-2037 clause 3.1, a new Certification Body (CB) test certificate shall be issued with a new CB test certificate number. Thanks to the fact that now the TD will have been revised to comply with technical state of the art, a smooth certification process will be assured.
Let´s focus on the introduction to IEC 60601-1-2 clause 8.11 – immunity to proximity magnetic fields in the frequency range 9 kHz to 13,56 MHz.
Subclause 8.11 explains why this requirement has been added. Due to a wide variety of sources with radiated fields in professional healthcare facility environments and home healthcare environments, concerns about risks have been raised. Many medical electrical equipments contain electronic components and circuitry which might be sensitive to radiated magnetic fields (source: Cf. IEC 60601-1-2 Edition 4.1 Subclause 8.11). Figure A.3 – Steps for evaluation of immunity to proximity magnetic fields provides a great overview if testing in accordance with Table 11 of IEC 60601-1-2 4.1 Edition is applicable. IEC 61000-4-39 electromagnetic compatibility – part 4-39: Testing and measurement techniques – radiated fields in close proximity – immunity test, describe the test and measurement condition.
Another noteworthy update of IEC 60601-1-2 Edition 4.1 affects clause 4.3.3 power input voltage and frequencies. Table 1 of clause 4.3.3 specifies the power input voltages and frequencies during testing. For instance, conducted disturbances (conducted emissions) according to CISPR11:2015+AMD1:2016+AMD2:2019 specifies the power input voltage for minimum and maximum rated voltage, if the difference between the maximum and the minimum rated input voltage is greater than 25% of the highest rated input voltage. Meaning, if your medical electrical equipment has a wide range input voltage supply of 100Vac – 240Vac, the difference of maximum and minimum rated input voltage is 140Vac, which is greater than 25% of highest rated input voltage (240Vac*25% = 60Vac), therefore the test needs to be conducted at minimum and maximum rated input voltage.
Previous IEC 60601-1-2 Edition 4.0 specified this test according to CISPR11 at any one voltage.
How can Medidee support your company?
Medidee has a dedicated team of electrical safety specialists who assist you in identifying all areas requiring updates to meet the new requirements introduced by IEC 60601-1-2 Edition 4.1, through a detailed review of the technical documentation of your medical electrical device or system, as well as provide support in the implementation of applicable new IEC 60601-1-2 Edition 4.1 requirements and sub-standards like CISPR 11 or IEC 61000-4-39. For instance, Medidee reviews and updates the technical documentation according to the updated standards to comply with IEC 60601-1-2 Edition 4.1. Furthermore, Medidee updates the test plan which is required as input for external test labs.
In addition, Medidee supports you in communicating with external accredited testing laboratories and notified bodies, to ensure a smooth certification process of your medical electrical equipment or system.
Contact Medidee today to discuss your needs and how Medidee supports you in addressing them: www.medidee.com/contacts
This article was written by Stefan Staltmayr.

[ARTICLE] Sampling Strategy: 10 FAQs on Sample Size Selection
Whether you are in the feasibility stage, verifying your design inputs and outputs, or validating your manufacturing process, choosing the wrong sample size for your tests can have a significant impact on your project, especially when this is detected during the market approval phase. Not only will it impact your time-to-market, but you will also likely find yourself repeating expensive testing, or worse, redesigning your product or process if you discover that adequate reliability is in fact not achieved.
Besides the benefits to your product development, sample size selection – including written justification – is also a regulatory requirement. Perhaps the most straightforward regulation is that from the FDA, found in the Quality System Regulation, 21 CFR §820.250(b), which states:
“Sampling plans, when used, shall be written and based on a valid statistical rationale. Each manufacturer shall establish and maintain procedures to ensure that sampling methods are adequate for their intended use... These activities shall be documented.”
Similarly, ISO 13458:2016 §7.3.6 and 7.3.7 state:
“The organization shall document verification … [and] validation plans that include methods, acceptance criteria and, as appropriate, statistical techniques with rationale for sample size.” These are clear requirements to include a written sample size justification in your testing protocols.
While the newly updated EU regulations (MDR/IVDR) do not carry explicit requirements like those of the FDA and in ISO 13485:2016, there is an implied requirement to provide a justification of your sample size selection through the requirements to show compliance with harmonized standards, found throughout the regulations. This includes ISO 13485:2016 in a very broad manner, but can also apply to topic-specific standards, such as those for sterilization or biocompatibility.
It is never too early in your project to start planning for sample size selection, considering its impact on your timelines, the cost of tests or the quality and quantity of representative devices to be produced to that end.
Here are 10 commonly asked questions we get when supporting clients with their sampling strategy.
1. Can’t I just use a sample size of 30 all the time?
First and foremost, the sample size you select should give you the information you need. Are you conducting an early feasibility test to inform your product design? Are you conducting design validation on a frozen design? These are very different types of tests which can require different sampling strategies. To get the most out of your testing – which can be costly to repeat – make sure to select an appropriate sample size to get the information you need. While n=30 may work for some tests, it will not always be the best sample size to meet your needs.
Besides type test (a test on one representative sample of the device with the objective of determining if the device, as designed and manufactured, can meet its requirements), some product-specific standards may include predefined sample size. In all the other cases, a statistical rationale shall be developed.
2. What exactly is a Confidence Level?
How do you know if your test results are meaningful? Choosing a sample size which is too small can leave the story of your testing incomplete – like trying to see the finished image of a puzzle without all the pieces in place. You can think of your confidence level like a 100-piece puzzle: how many pieces of the puzzle do you need to correctly guess the image? The more pieces you have, the more sure you can be about what the image is. The same is true for confidence level – a higher percentage will give you greater assurance that your test results are correct.
3. How do I pick the right Confidence Level?
To determine your confidence level, you need to understand the impact of your results being wrong in order to understand the importance of your results being correct. This is often tied to the product development stage your testing is being conducted in, as well as the risk of “false positives”. Another practical concern is the cost of samples and the time to produce them. While this is an important consideration for your overall project – it cannot be the driving factor and should instead be a secondary consideration when determining sample size. While a low confidence level may seem appealing due to the lower number of required samples, the result may not give you the information you need.
4. Do I need to select a Reliability too?
In short, for design verification, yes. Reliability is a measure of how well a device or product will perform under a certain set of conditions for a specified amount of time. It is an indication of the consistency of the measurement and is critical to specify when selecting a sample size. Like selecting a confidence level, the reliability is related to the risk of the test results being incorrect. Reliability criteria which is too low – leading to a smaller sample size – may lead to test results which do not appropriately reflect the acceptable failure rate of your device.
5. Does it matter what type of test I’m conducting?
The type of test can impact the sample size. It is important to consider if your samples will undergo cyclic testing, if they require real-time or accelerated aging, if the samples will be tested to failure, and even if they need to be representative of different lots, shifts, or manufacturing sites. All of these details and more can inform your sample size selection and how the sample size is defined. For example, if a device is produced at two separate manufacturing sites, careful consideration must be taken to determine if the sites should be sampled together or separately. A similar grouping determination should be made with samples that go through cleaning, sterilization, or aging.
6. Do I need to link my sample size to my risk assessment?
As already discussed, your sample size selection must be informed by the risk of your test results being incorrect or incomplete. However, it should also be tied to the device risks. A pre-determined matrix connecting risk levels and confidence and reliability can add consistency into your sampling process. More severe risks with a higher probability of occurring generally require a higher confidence and reliability level and therefore a higher sample size. Linking these in your sampling procedure can help define your sample size justification.
7. How do I determine which risks are impacted?
In order to link your sample size to your risk assessment, you must determine which risks are related to the test you are conducting. First, identify what the test is challenging – is it a design validation test which is intended to confirm a specific feature of the design? Or perhaps a process validation which will serve in place of an inspection step? In some cases, the failure of the test itself will be directly linked to a risk in your risk analysis. In other cases, the resulting consequence of the failure may be the related risk. In your justification, you can reference the risks which are impacted.
8. Does this approach work for all data types?
The type of data you will receive from your test may help to determine your sample size. Will your test results be pass/fail (or accept/reject)? Will you be testing a numerical value against a one- or two-sided specification? Are you conducting a comparative test between two samples? Depending on the type of data, the method for choosing a sample size may be limited to only one of several different methods and can help you rule out other possible sample size selection processes.
9. How many samples can “fail” and still have an acceptable test?
Now that you’ve determined your confidence and reliability levels, type of test, impacted risks, and data type, the last key parameter to determine is the number of allowable failures. In many cases, the standard is to allow for no failures, or the “c=0” sampling approach. However, this is not the only way to do this. In some cases, particularly depending on the associated risks, it may be acceptable to use a threshold of allowable failures greater than 0. For these situations, it is important to define this in the justification.
10. Any tips on how to reduce my sample size?
Using a statistical approach doesn’t mean you can’t get pragmatic with your sampling plan! There is often more than one way to solve the riddle of sample size selection, with varying results. It can be helpful to talk through testing plans with a cross-functional group to determine the testing – and sample size – which meets your project needs while still using a statistically valid approach. This group can include quality engineers, product designers, manufacturing personnel, and materials experts. The sample size you select must be statistically justified, but that doesn’t mean the methodology, and therefore the answer, is always the same. If your first selection is unreasonably high – for example, you need to test more samples than you produce annually – then go back to the drawing board and think outside the box.
As you can see, developing an adequate sampling strategy requires a strong set of skills, including knowledge of your product, statistics and risk management, all combined with pragmatism and cleverness.
With extensive track record working on similar problematics, Medidee can support you with services ranging from training courses and coaching, up to complete preparation of your sampling strategy.
Contact us today to discuss your project!
This article was written by Paige Elizabeth Sutton.

[WEBINAR] Medical Devices Incorporating or Generating Nanomaterials
Medical Devices Incorporating or Generating Nanomaterials
The concept of nanomaterials and their possible applications was first introduced by Richard Feynman, which earned him the Nobel Prize in 1959. Fast-forward to today, nanomaterials have permeated all aspects of our daily lives, including their utilization in medical devices. Offering the potential to enhance device functionality and diagnostic outcomes, these tiny particles (10⁻⁹ meters) have the power to revolutionize healthcare.
However, their application in medical devices presents a unique challenge: ensuring their safety and compatibility with the human body, within a context of lack of standardized testing protocols, limited available literature data, and uncertainty regarding exposure, among other factors. This is crucial because nanomaterials can have sizes similar to structures at the subcellular level, and thus can interact with such structures, presenting serious biological risks.
In this recorded webinar, Dr Neeru Mittal and Dr Jonathan Kelly from Veranex provide you with:
- A basic understanding of biocompatibility
- An introduction to the diverse array of nanomaterial types and their applications within the realm of medical devices
- Principles of biological safety evaluation of medical devices incorporating or generating nanoparticles
WATCH THE WEBINAR
Please submit the form to watch:

[WEBINAR] Leveraging FDA Clearance/Approval for Successful EU MDR Submissions
LEVERAGING FDA CLEARANCE/APPROVAL FOR SUCCESSFUL EU MDR SUBMISSIONS
The transition phase to MDR (Regulation (EU) 2017/745) has been underway since mid-2020 and has introduced new requirements for medical devices.
Experience gained with numerous Notified Bodies during this critical time has highlighted common pitfalls and areas where medical device companies face the greatest challenges. However, it has also shown how companies who have successfully navigated the FDA can use this to their advantage in the CE marking process.
In this recorded webinar, Kristi Nakayama, Antonia Claasz and Paige Sutton-Smith from Veranex guide you through some of the most important aspects you should consider for your MDR submission, including:
- Overview of requirements of CE marking per the MDR
- Potential gaps to be bridged
- Advantages of previous FDA clearance/approval
WATCH THE WEBINAR
Please submit the form to watch:

[TECH LETTER] Medical Device Software incorporating Artificial Intelligence: Generating sufficient evidence under the MDR
Artificial Intelligence (AI) and Machine Learning (ML) technologies have the potential to transform medicine by aiding in the detection, diagnosis, and management of diseases. As digitalization of healthcare generates massive amounts of data, medical device manufacturers are increasingly incorporating AI technologies to automate the analysis of such data targeting to create innovative products and improve patient care. This turn towards AI-enabled medical device software (MDSW) is also evidenced by the plethora of studies evaluating the feasibility of artificial intelligence systems across a wide range of health-related indications.
While the interest in medical applications of AI is strong, inconsistent and incomplete collection of evidence remains one of the barriers to the assessment of the safety and performance of AI-MDSW by regulatory bodies.
According to the provisions of Article 61(1) of the MDR EU 2017/745, it is the responsibility of the manufacturer to specify and justify the level of Clinical Evidence necessary to demonstrate conformity of their medical device to the relevant General Safety and Performance Requirements (GSPRs); this level of clinical evidence should be appropriate in view of the device characteristics and intended purpose.
Determining the appropriate level of evidence might be challenging, especially in the case of AI-enabled MDSW which significantly differs from established medical device software in terms of technical and clinical aspects. At the same time, there is no explicit regulatory guidance for conformity assessment of AI technologies, delineating appropriate and practical evidence generation approaches.
Accordingly, this Technical Letter aims to provide an overview of the considerations for evaluating evidence regarding AI-MDSW.
GET THE TECH LETTER
Please submit the form: